- Overview
- Epidémiologie
- Classification biochimique et nomenclature
- Génétique
- Pathophysiologie
- Défauts de glycosylation des protéines liées à l’azote
- Défauts de glycosylation liée à l’O et défauts combinés de glycosylation liée à l’N et à l’O
- Glycosylation des lipides et défauts de biosynthèse des ancres GPI
- Manifestations cliniques
- Défauts de glycosylation des protéines N-liées
- PMM2-CDG (CDG-Ia, déficit en PMM2)
- MPI-CDG (CDG-Ib, déficit en mannosephosphate isomérase)
- ALG6-CDG (déficit en glucosyltransférase 1)
- Défauts de glycosylation liés à l’oxygène et défauts combinés de glycosylation liés à l’azote et à l’oxygène
- Défauts de glycosylation des lipides et de biosynthèse des ancres GPI
- Diagnostic
- Défauts de glycosylation des protéines liées à l’azote
- Défauts de glycosylation O-liée et défauts de glycosylation N- et O-liée combinés
- Défauts de biosynthèse de la glycosylation des lipides et des ancres GPI
- Analyse moléculaire
- Gestion
- Thérapies ciblées et pronostic
- Remerciements
- Note de bas de page
Overview
La glycosylation est le processus d’ajout de résidus de sucre aux protéines et aux lipides dans différentes voies cellulaires. Les troubles congénitaux de la glycosylation (TCG) constituent un groupe génétiquement et cliniquement hétérogène de plus d’une centaine de maladies causées par des défauts dans diverses étapes des voies de synthèse ou de modification des glycanes. La plupart de ces maladies monogéniques ont un mode d’hérédité autosomique récessif, mais des formes autosomiques dominantes et liées au chromosome X ont également été décrites.
Les TGC se manifestent généralement par des manifestations multisystémiques, le plus souvent un retard de développement, un retard de croissance, une hypotonie, des anomalies neurologiques, une hépatopathie et une coagulopathie. Les personnes touchées peuvent également présenter des maladies oculaires, cutanées et cardiaques, ainsi que des dysmorphismes faciaux. Bien que des changements neurologiques et des retards cognitifs soient observés chez la majorité des individus affectés, il existe certains cas et même des types qui ne présentent pas de manifestations neurologiques. Compte tenu de la large étiologie clinique et génétique de la GDC, le diagnostic clinique repose sur un indice de suspicion élevé dans le cas d’une maladie multisystémique.
L’analyse de la transferrine déficiente en glucides (CDT) dans le sérum est le test de dépistage de première ligne chez les patients suspectés de GDC, mais sa détection est limitée aux défauts de N-glycosylation avec des déficiences en acide sialique. Les tests suivants comprennent l’analyse des glycanes liés au dolichol et les tests génétiques. Le diagnostic précoce de ce groupe de maladies à croissance exponentielle est important, car certaines GDC sont traitables. Le traitement des défauts de glycosylation est principalement un traitement de soutien, bien que des thérapies ciblées soient disponibles pour le MPI-CDG, le SLC35C1-CDG, le PIGM-CDG et le PGM1-CDG. Des détails sur ces traitements figurent dans la section « Thérapies ciblées et pronostic » ci-dessous. Cette revue se concentrera sur les types les plus courants de GDC avec des phénotypes ou des traitements reconnaissables, le public cible étant les prestataires de soins primaires.
Epidémiologie
L’incidence et la prévalence de tous les types de GDC dans l’ensemble n’ont pas été bien établies, bien que des patients aient été signalés dans le monde entier dans presque toutes les origines ethniques et que les deux sexes soient également touchés. La prévalence estimée dans les populations européennes et afro-américaines est de 1/10 000 sur la base des fréquences de portage des variants pathogènes connus dans 53 gènes (1-4). La prévalence de la CDG la plus fréquemment diagnostiquée, la PMM2-CDG, varie de 1/20 000 dans les populations néerlandaises à 1/77 000 en Estonie sur la base de rapports isolés (5,6). A ce jour, moins de 100 cas ont été rapportés pour la plupart des types de CDG.
Classification biochimique et nomenclature
De manière générale, les CDG sont actuellement classés en quatre catégories-(I) glycosylation N-liée, (II) glycosylation O-liée, (III) glycosylation combinée N- et O-liée/multiple, et (IV) défauts de biosynthèse des lipides et des ancres glycosylphosphatidylinositol (GPI).
Le défaut de glycosylation des protéines N-liées PMM2-CDG, précédemment connu sous le nom de CDG de type Ia, a été le premier CDG à être signalé par Jaeken en 1980 et reste de loin le CDG le plus courant à ce jour (7). Le PMM2-CDG a été initialement appelé « syndrome de la glycoprotéine déficiente en hydrates de carbone » en raison des multiples anomalies des glycoprotéines sériques observées par focalisation isoélectrique de la transferrine sérique chez les individus affectés. Historiquement, les GDC ont été classés en fonction des schémas d’analyse des isoformes de la transferrine – les schémas de type I étaient attribués à des défauts d’assemblage et de transfert de glycanes liés aux dolichols, localisés dans le cytoplasme ou le RE, et les schémas de type II étaient attribués à des défauts de traitement dans l’appareil de Golgi. À partir de ce point d’embranchement, les GDC ont ensuite été nommés alphabétiquement dans l’ordre de leur découverte.
Avec l’avènement du diagnostic moléculaire généralisé, la nomenclature des GDC a été mise à jour en 2008 pour spécifier l’étiologie moléculaire de la maladie, reflétant la croissance exponentielle des voies et des troubles qui ne rentraient pas dans les catégories dichotomiques précédemment établies. Actuellement, la nomenclature des GDC est désignée par le nom du gène affecté (non italique, noms de gènes à www.genenames.org), suivi de -CDG (par exemple, PMM2-CDG) (8).
Génétique
La grande majorité des troubles congénitaux de la glycosylation sont hérités sur un mode autosomique récessif, avec une mutation héritée de chaque parent asymptomatique (porteur). Des tests moléculaires, généralement avec des méthodes de séquençage de nouvelle génération, sont nécessaires pour établir un diagnostic génétique. Les tests parentaux pour la variante connue peuvent confirmer l’hérédité ou l’apparition de novo. Pour l’hérédité autosomique récessive, le risque de récurrence pour les frères et sœurs et chaque grossesse d’un individu affecté est de 25% pour être affecté, 50% pour être un porteur asymptomatique, et 25% pour ne pas être affecté.
Une poignée de CDG ont une hérédité autosomique dominante (N-liée : GANAB-CDG, PRKCSH-CDG ; O-liée : EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Un plus petit nombre est lié à l’X (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). La plupart des formes dominantes et certaines formes liées au chromosome X de la CDG sont dues à des mutations de novo. Les maladies et les gènes spécifiques sont décrits ci-dessous dans la section « physiopathologie ».
Les données de mutation pour tous les gènes publiés pour la CDG sont disponibles sur la base de données des mutations génétiques humaines (http://www.hgmd.cf.ac.uk/ac/index.php). Des informations sur les variantes de gènes spécifiques sont disponibles sur la base de données Leiden Open Variation Database avec des outils intégrés de pathogénicité in silico (http://www.lovd.nl/3.0/home). Des synopsis cliniques pour des gènes spécifiques sont disponibles sur le site Online Mendelian Inheritance in Man (http://www.omim.org/) ou, de manière plus limitée, sur GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Compte tenu du petit nombre de patients affectés pour la plupart des sous-types de GDC, la corrélation génotype-phénotype est difficile à établir.
Pathophysiologie
Plus de 130 types de GDC ont été rapportés à ce jour (9,10). Compte tenu de l’omniprésence des voies de glycosylation, les GDC sont extrêmement diversifiés dans leur pathogénie biochimique. De nombreuses protéines et lipides (c’est-à-dire les sphingolipides et les glycolipides) subissent une glycosylation avec des monosaccharides et/ou des oligosaccharides, collectivement appelés glycanes, dans différents compartiments cellulaires. Leurs localisations subcellulaires sont diverses, mais la plupart des défauts se produisent dans le RE ou l’appareil de Golgi. Les caractéristiques cliniques et l’étiologie génétique des GDC les plus courants par voie sont résumées dans le tableau S1.
Parmi les protéines, les glycanes sont décrits par leur liaison à la chaîne polypeptidique – les N-glycanes sont attachés au groupe amide de l’asparagine (Asn) tandis que les O-glycanes sont attachés au groupe hydroxyle de la sérine ou de la thréonine. La synthèse des N-glycanes nécessite la construction par étapes de sucres liés à des nucléotides dans le cytosol, l’assemblage dans le réticulum endoplasmique et la transformation dans l’appareil de Golgi. En revanche, la synthèse des O-glycanes nécessite un assemblage mais pas de traitement, donc les défauts de O-glycosylation se produisent principalement dans l’appareil de Golgi.
Défauts de glycosylation des protéines liées à l’azote
La N-glycosylation implique la fixation covalente de structures glucidiques au groupe amide de la chaîne latérale des résidus Asn au sein d’un site accepteur consensus Asn-X-Ser/Thr, la translocation du polypeptide substrat vers le réticulum endoplasmique pour le remodelage, et une modification supplémentaire de la chaîne N-glycane au sein du Golgi (11,12). Des défauts à n’importe quel endroit de la voie de synthèse, d’assemblage et de transformation peuvent entraîner une maladie clinique.
La PMM2-CDG est causée par des variantes pathogènes du gène de la phosphomannomutase 2 (PMM2), entraînant une déficience de l’enzyme PMM2 qui catalyse la conversion cytosolique du mannose-6-phosphate en mannose-1-phosphate lors de la deuxième étape de la synthèse du mannose par le guanosine diphosphate (GDP). La plupart des patients sont porteurs de mutations missens pathogènes hétérozygotes composées (www.lovd.nl/PMM2). La variante pathogène récurrente la plus courante, p.Arg141His, est présente chez environ 40 % des personnes atteintes d’ascendance européenne, et p.Phe119Leu est également fréquemment trouvée en Europe du Nord (1). Des corrélations génotype-phénotype ont été rapportées pour PMM2-CDG (3,13,14).
MPI-CDG est une maladie autosomique récessive causée par des variants pathogènes dans le gène de la mannose phosphate isomérase (MPI) conduisant à une phosphomannose isomérase (MPI) déficiente. La MPI catalyse normalement la première étape de la synthèse du GDP-mannose (c’est-à-dire la conversion du fructose-6-phosphate en mannose-6-phosphate), mais le fructose-6-phosphate ne s’accumule pas dans les cellules car il peut également être métabolisé par la voie glycolytique. Par conséquent, bien qu’il soit biochimiquement similaire au PMM2-CDG, le MPI-CDG ne provoque pas d’atteinte neurologique et multisystémique aussi importante. Le CDT est également le test de dépistage de choix pour le MPI-CDG, qui présente un profil de type 1. Le diagnostic peut ensuite être confirmé par voie moléculaire ou par l’activité MPI des fibroblastes/leucocytes.
L’ALG6-CDG est une maladie récessive causée par des mutations de l’ALG6, entraînant une fixation anormale de trois molécules de glucose sur des intermédiaires de mannose liés au dolichol et une hypoglycosylation en aval des glycoprotéines sériques (15).
Défauts de glycosylation liée à l’O et défauts combinés de glycosylation liée à l’N et à l’O
La glycosylation liée à l’O comprend l’ajout par étapes de chaînes d’hydrates de carbone aux résidus sérine, thréonine et hydroxylysine des protéines par des glycosyltransférases dans l’appareil de Golgi (16). Plusieurs types de glycanes O-liés ont été associés à des maladies humaines, nommés par le premier sucre attaché au résidu d’acide aminé (17).
Glycosylation des lipides et défauts de biosynthèse des ancres GPI
Les ancres GPI sont des glycolipides qui subissent un assemblage séquentiel dans le réticulum endoplasmique et des modifications dans le Golgi. Les troubles de la biosynthèse des ancres GPI dus à des déficiences enzymatiques sont nommés par ordre alphabétique de découverte et non chronologiquement par étape d’assemblage. Une fois synthétisées, les ancres GPI résident sur les membranes plasmatiques et se lient à des centaines de protéines de surface des cellules, remplissant une pléthore de fonctions cellulaires. La plupart de ces maladies sont autosomiques récessives à l’exception notable du déficit en PIGA lié au chromosome X.
Manifestations cliniques
En raison de l’omniprésence des voies de glycosylation, pratiquement tout système organique peut être impliqué dans la GDC, bien que la plupart des cas impliquent des anomalies neurologiques. Certains CDG se présentent avec une ichtyose, notamment MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (figure 1) et PIGL-CDG (18,19). Presque tous les CDG présentent une maladie multisystémique au cours des premières années de vie, sauf certains qui n’affectent qu’un seul système organique (ex, la rétine avec DHDDS-CDG ; la jonction neuromusculaire avec ALG2-CDG, ALG14-CDG, CFPT1-CDG ; le cerveau avec ST3GAL3-CDG, TUSC3-CDG ; la peau ou les muscles squelettiques avec POGLUT1-CDG, POFUT1-CDG ; le cartilage avec EXT1/EXT2-CDG ; le foie avec TMEM199-CDG ; les globules rouges avec SEC23B-CDG). L’âge d’apparition et la gravité peuvent aller de la létalité néonatale à l’âge adulte presque asymptomatique, et toute permutation entre les deux. La constellation de symptômes la plus fréquemment signalée comprend un retard de développement, un retard de croissance, une hypotonie, des anomalies neurologiques, une hypoglycémie et des anomalies variables du foie, des yeux, de la peau, du système gastro-intestinal, de l’immunologie, du squelette et de la coagulation (19).
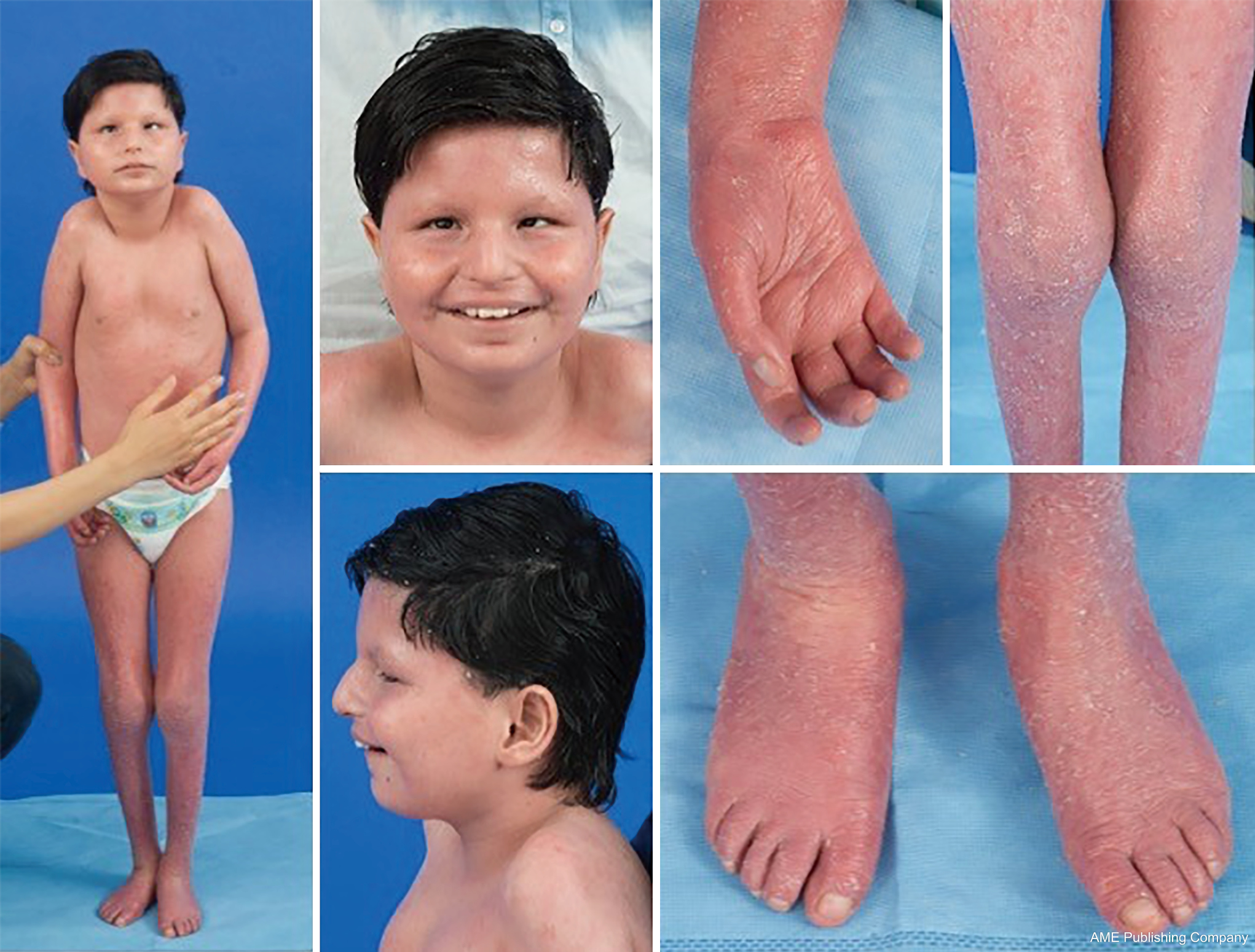
Le phénotype complet de nombreux sous-types de CDG reste à délimiter complètement en raison de la rareté des cas rapportés. Par conséquent, la GDC doit être envisagée dans tout contexte de maladie multisystémique, en particulier dans les cas présentant une composante neurologique ou un retard de développement non spécifique dont l’étiologie n’est pas claire.
Bien que la physiopathologie des symptômes de la multitude reste à élucider, la relation entre certaines voies de glycosylation et des symptômes cliniques spécifiques a été clarifiée. Par exemple, le retard de croissance observé dans de nombreux types de GDC est attribuable à l’hypoglycosylation et à l’altération de la formation de plusieurs glycoprotéines dans la voie de croissance de l’insuline, notamment l’IGF-1, l’ALS et l’IGFBP-3 (20). À mesure que nous comprenons mieux ce groupe de troubles complexes, les GDC sont de plus en plus reconnus chez les personnes dont le diagnostic est difficile à établir. Les systèmes d’organes impliqués dans les différents GDC sont résumés dans le tableau S1. Nous discuterons des caractéristiques cliniques des formes les plus courantes et des formes avec des traitements ciblés de CDG ci-dessous.
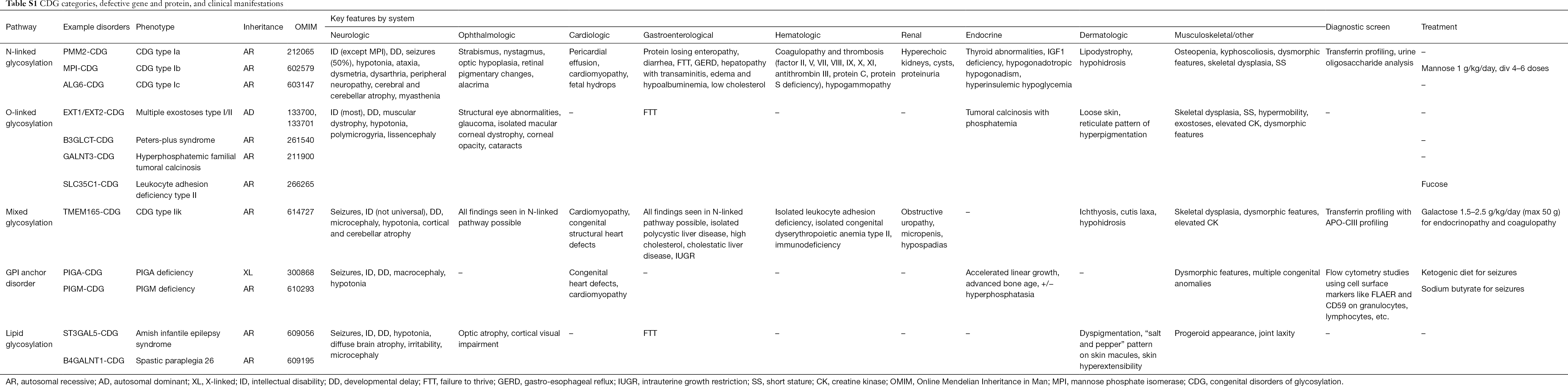
Tableau complet
Défauts de glycosylation des protéines N-liées
En tant que CDG le plus fréquemment diagnostiqué, le phénotype des troubles de la glycosylation N-liée est souvent annoncé comme la présentation classique. Cependant, le spectre phénotypique des CDG est très diversifié, et de nombreux CDG peuvent ne pas présenter les symptômes stéréotypés associés à la PMM2-CDG.
PMM2-CDG (CDG-Ia, déficit en PMM2)
La PMM2-CDG est la CDG la plus courante, avec plus de 700 cas rapportés dans le monde. Il se caractérise par une maladie grave multisystémique dans la petite enfance, une maladie neurologique et un retard de développement dans l’enfance, et/ou une déficience intellectuelle stable à l’âge adulte (21,22).
Dans la petite enfance, le PMM2-CDG présente des anomalies neurologiques typiques peu après la naissance, à savoir un strabisme et des mouvements oculaires anormaux, une hypoplasie cérébelleuse, une hypotonie, un retard psychomoteur, une ataxie, une hypotonie et une hyporéflexie. Les nourrissons peuvent également souffrir d’une maladie hépatique, d’un syndrome néphrotique et de kystes rénaux, d’un épanchement péricardique et d’une cardiomyopathie hypertrophique, d’un retard de croissance et d’une défaillance multi-organique entraînant la mort au cours de la première année de vie chez jusqu’à 20 % des individus affectés (21,23-28).
Une constellation de caractéristiques dysmorphiques a été décrite chez les patients atteints de PMM2-CDG (figures 2,3). Il s’agit notamment d’un cervelet hypoplasique, de dysmorphismes faciaux (c’est-à-dire de grandes oreilles dysplasiques), de mamelons inversés et d’une répartition anormale du tissu adipeux sur les fesses ou la région sus-pubienne qui peut se résorber avec l’âge (14,21,29-32). Les patients ont été décrits comme ayant un comportement extraverti et heureux. La présentation est très variable, bien que le strabisme puisse être observé chez plus de 70 % des patients affectés (21,23,33-35). Des mamelons inversés et des coussinets graisseux anormaux sont observés chez environ 25-50% des patients (36).

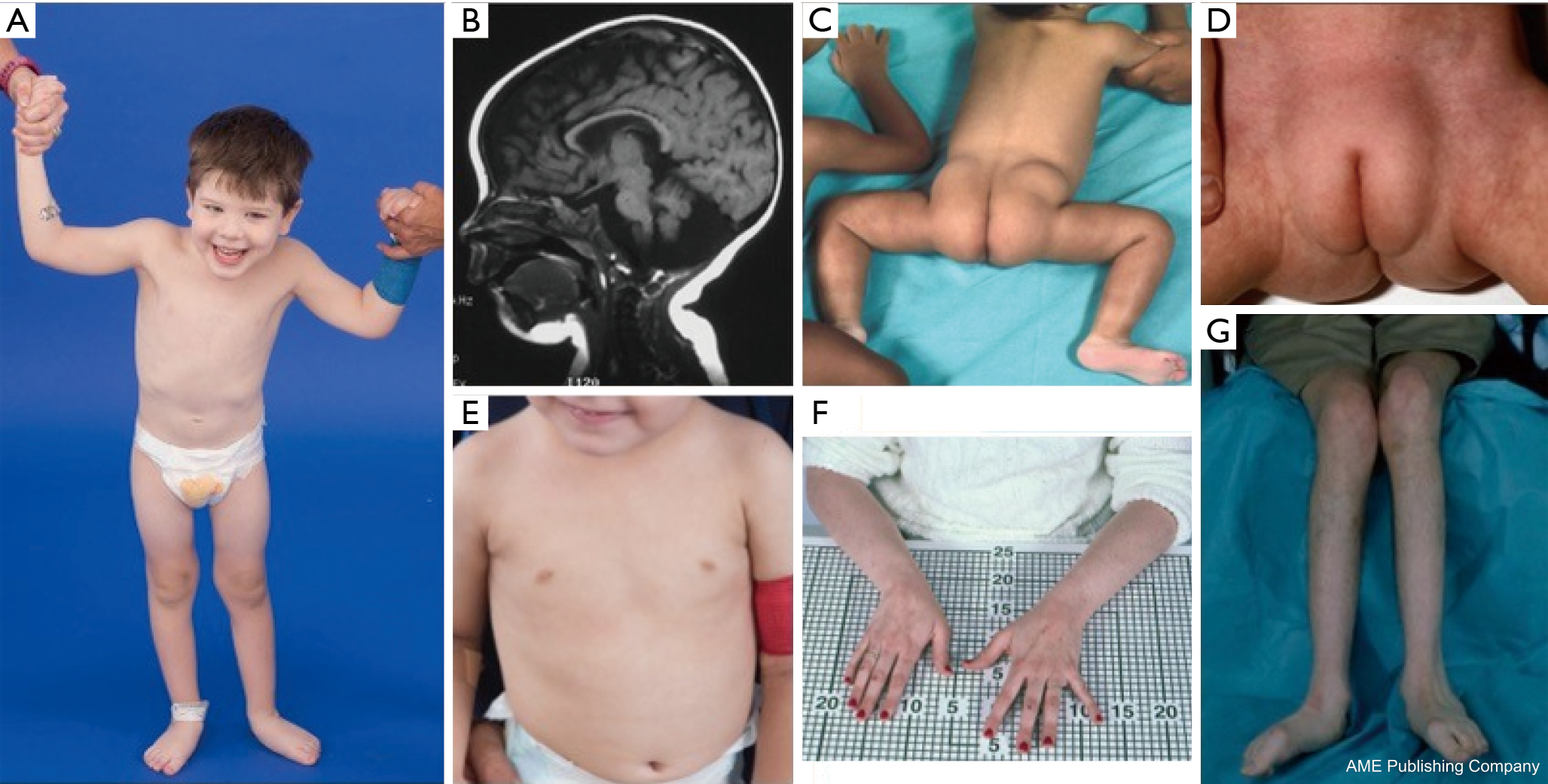
Dans l’enfance, les personnes atteintes peuvent développer une rétinite pigmentaire, des épisodes de type AVC et des crises d’épilepsie, des retards de parole et de motricité, et une neuropathie périphérique. Sur le plan constitutionnel, les patients présentent fréquemment un retard de croissance dû à des anomalies alimentaires et gastro-intestinales et un retard global de développement. On peut observer des transaminases hépatiques élevées sans conséquence clinique, qui se normalisent généralement vers l’âge de 5 ans avec des fluctuations occasionnelles en fonction de la maladie (21,24). Les biopsies hépatiques sont rarement indiquées en cas de GDC, sauf si l’on soupçonne une fibrose hépatique (1). L’hypothyroïdie clinique est rare, mais les patients atteints de GDC doivent faire mesurer leurs hormones thyroïdiennes et leur T4 libre, ce qui peut montrer une faible globuline liant la thyroïde (TBG) et des élévations transitoires de l’hormone stimulant la thyroïde (TSH) (37). Les malformations du foie et des voies biliaires n’ont pas été rapportées chez les patients atteints de PMM2-CDG.
Les adultes atteints de PMM2-CDG peuvent vivre jusqu’à leur 7e ou 8e décennie avec un retard cognitif stable, une neuropathie périphérique et une cyphoscoliose thoracique et spinale progressive avec ostéopénie ou ostéoporose (34). L’ataxie cérébelleuse est un symptôme de plus en plus reconnu avec une atteinte multisystémique (38-40). Les anomalies endocriniennes comprennent l’hyperprolactinémie, la libération d’hormone de croissance avec hyperglycémie, la résistance à l’insuline et l’hypoglycémie hyperinsulinémique (41,42). Chez les femmes atteintes, l’hypogonadisme hypogonadotrope peut entraîner l’absence de développement sexuel secondaire ou l’absence d’ovaires (41,43,44). Les patients peuvent présenter un risque accru de thrombose en raison de la diminution des facteurs de coagulation sériques, notamment les facteurs IV, IX et XI, l’antithrombine III, la protéine C et la protéine S (29).
MPI-CDG (CDG-Ib, déficit en mannosephosphate isomérase)
MPI-CDG est unique car les patients atteints ont peu ou pas d’atteinte neurologique et certaines manifestations de la maladie sont traitables par le mannose oral (2). Les symptômes sont principalement hépatiques-intestinaux sans caractéristiques dysmorphiques ou retards cognitifs. Les patients présentent généralement des vomissements récurrents, une hypoglycémie importante, un retard de croissance, une entéropathie protéique potentiellement mortelle, des modifications fibrotiques du foie et une dilatation des voies biliaires (45-51). Les patients présentent un risque accru d’événements thrombotiques en raison des faibles concentrations sériques de protéines C et S et d’antithrombine III.
ALG6-CDG (déficit en glucosyltransférase 1)
L’ALG6-CDG est le deuxième défaut de N-glycosylation le plus fréquent caractérisé par un phénotype similaire mais plus léger que le PMM2-CDG. Les patients atteints d’ALG6-CDG présentent un retard de croissance, un retard de développement, une hypotonie, des crises d’épilepsie, un strabisme, une ataxie, une coagulopathie et des dysmorphismes faciaux (c’est-à-dire des oreilles décollées, un hypertélorisme et une macroglossie). Comme pour le MPI-CDG, ils peuvent également présenter une entéropathie avec perte de protéines. En outre, les patients atteints peuvent présenter des anomalies squelettiques, notamment une brachydactylie, des malformations des doigts et une scoliose. Les patients atteints ne présentent généralement pas de rétinite pigmentaire ou d’hypoplasie cérébelleuse (52).
Défauts de glycosylation liés à l’oxygène et défauts combinés de glycosylation liés à l’azote et à l’oxygène
En raison de la présence substantielle de O-glycanes dans les protéines contenant de la mucine, y compris les glycosaminoglycanes (GAG) et les surfaces épithéliales (53), les troubles de la synthèse des GAG entraînent généralement des dysplasies du squelette ou des maladies du tissu conjonctif. Les patients atteints peuvent présenter des anomalies musculo-squelettiques, cutanées et articulaires (par exemple, laxité articulaire, exostoses multiples, chondro/ostéosarcomes) en plus de symptômes neurologiques (54-56). Par exemple, la N-acétylgalactosaminyltransférase 3 (GALNT3) O-glycosyle l’hormone phosphaturique, FGF23, empêchant le clivage protéolytique et permettant sa sécrétion intacte. La déficience en GALNT3 entraîne une calcinose tumorale familiale, caractérisée par une hyperphosphatémie et des calcifications ectopiques (57,58).
Défauts de glycosylation des lipides et de biosynthèse des ancres GPI
Les glycosphingolipides et leurs dérivés sialylés, les gangliosides, sont principalement exprimés par les neurones. Les défauts dans la dégradation des gangliosides entraînent une accumulation et les maladies de stockage lysosomales bien caractérisées. À l’opposé, les défauts dans la biosynthèse des gangliosides, tels que ST3GAL5-CDG et B4GALNT1-CDG, sont extrêmement rares et entraînent des maladies neurodégénératives graves. Les patients peuvent présenter une paraplégie spastique, un retard intellectuel sévère, une épilepsie et des symptômes non neurologiques, notamment une dysplasie squelettique, des caractéristiques dysmorphiques et une pigmentation cutanée anormale (59,60).
Les mutations de nombreux gènes au sein de la voie de biosynthèse des ancres GPI provoquent une variété d’anomalies congénitales multiples, une déficience intellectuelle et une épilepsie. Le défaut de biosynthèse GPI le mieux caractérisé, le déficit PIGA lié à l’X, se présente sous la forme de spasmes infantiles avec hypsarythmie, hypotonie, anomalies cérébrales multiples et dysmorphismes faciaux. Les patients peuvent également présenter des maladies variables de la peau, du foie, du cœur et des reins (61-69). Certaines mutations au sein de la PIGA provoquent la maladie phénotypiquement distincte de l’hémoglobinurie paroxystique nocturne (PNH), un trouble acquis de l’insuffisance de la moelle osseuse (70,71).
Diagnostic
Lorsqu’un GDC est cliniquement suspecté, la première étape consiste à demander des tests biochimiques de GDC dans le plasma ou le sérum, y compris des tests de CDT et de N-glycanes. L’analyse de la CDT et des N-glycanes sériques ne peut détecter que les défauts de N-glycosylation, elle ne serait donc pas utile pour différencier les défauts isolés de O-glycosylation ou d’ancrage GPI. L’analyse des isoformes de la transferrine était à l’origine obtenue par focalisation isoélectrique de la transferrine, car l’échec de la synthèse des N-glycanes entraîne une déficience partielle en acide sialique, ce qui modifie la charge de la transferrine sérique et par conséquent sa migration cathodique sur un champ électrophorétique. Cependant, l’analyse de la transferrine et du N-glycane par spectrométrie de masse a maintenant largement remplacé la focalisation isoélectrique en identifiant les changements spécifiques des oligosaccharides par la masse et la charge (72).
Défauts de glycosylation des protéines liées à l’azote
Les résultats du CDT de la transferrine sérique sont rapportés au ratio mono-oligosaccharide/di-oligosaccharide transferrine, a-oligosaccharide/di-oligosaccharide transferrine, tri-sialo/di-oligosaccharide transferrine, apolipoprotéine CIII-1/apolipoprotéine CIII-2, et apolipoprotéine CIII-0/apolipoprotéine CIII-2. Ces résultats quantitatifs s’accompagneront également d’une interprétation du schéma des résultats.
Un schéma de type I CDT de la transferrine est caractérisé par une augmentation des bandes de di- et d’asialotransferrine, et indique des défauts de synthèse des N-glycanes dans le cytosol ou le réticulum endoplasmique. Un modèle de type II est caractérisé par une augmentation des bandes de di- et d’asialotransferrine, et des bandes de tri- et/ou monosialotransferrine, et indique des défauts dans la transformation des N-glycanes dans l’appareil de Golgi (73).
Si un modèle de CDT de la transferrine sérique de type I est détecté, le déficit en PMM2 ou le déficit en MPI doit être au premier plan des différentiels, car le PMM2-CDG est le CDG le plus commun et le MPI-CDG est traitable et potentiellement fatal s’il n’est pas traité. Pour différencier les diagnostics, il convient d’effectuer un profilage des N-glycanes, un séquençage moléculaire ou des tests enzymatiques. Le diagnostic de PMM2-CDG ou MPI-CDG est confirmé par des tests moléculaires montrant des variants pathogènes bialéliques dans la PMM2 ou la MPI, puis par l’activité enzymatique de la PMM ou de la MPI dans les leucocytes ou les fibroblastes si la pathogénicité des variants génétiques est incertaine. L’analyse des N-glycanes ou l’analyse moléculaire permettrait de différencier la majorité des ALG-CDG des PMM2 ou MPI-CDG (15).
Un schéma CDT de la transferrine sérique de type II indique des défauts du Golgi tels qu’un déficit en N-acétylglucosaminyltransférase (GnT) II (CDG de type IIA, MGAT2-CDG). L’analyse de l’isoforme de l’apolipoprotéine CIII (Apo-CIII) est un test complémentaire pour un profil CDT de type II, car elle mesure les défauts de O-glycosylation des mucines dans l’appareil de Golgi. La sensibilité de la CDT ou de l’Apo-CIII pour la détection de la GDC de type II est limitée. Ainsi, le profilage des N-glycanes et des O-glycanes et le séquençage du panel moléculaire ou de l’exome doivent être entrepris lorsque ces tests cliniques sont disponibles. Les profils de glycosylation de la transferrine peuvent se normaliser de manière sporadique ; par conséquent, des tests répétés peuvent être indiqués chez les patients présentant un indice de suspicion élevé. Des faux positifs peuvent être obtenus chez les patients présentant une crise aiguë d’intolérance héréditaire au fructose, une galactosémie, une maladie hépatique aiguë et certaines infections bactériennes. Aucun des tests biochimiques de dépistage des GDC ne peut dépister tous les GDC. Ainsi, même en présence de résultats de dépistage normaux, un test de panel de gènes moléculaires ou un séquençage de l’exome peuvent être réalisés en cas de forte suspicion clinique. Inversement, la confirmation biochimique et fonctionnelle des résultats de la génétique moléculaire est également essentielle, car la majorité des patients atteints de GDC sont porteurs d’au moins une mutation missense légère et souvent nouvelle.
Défauts de glycosylation O-liée et défauts de glycosylation N- et O-liée combinés
Le diagnostic repose sur le séquençage moléculaire, car l’analyse des isoformes de la transferrine ne permettrait pas de détecter les défauts de O-glycosylation isolés. Les défauts combinés de glycosylation liée à la N et à la O peuvent être détectés par la CDT, l’analyse de l’ApoCIII et l’analyse des N-glycanes et O-glycanes plasmatiques.
Défauts de biosynthèse de la glycosylation des lipides et des ancres GPI
La cytométrie en flux des granulocytes sanguins mesure l’expression à la surface des cellules des protéines à ancres GPI telles que CD16 et CD24. L’analyse par cytométrie en flux des globules blancs ou des globules rouges pour certaines protéines de surface cellulaire ancrées sur GPI sont disponibles cliniquement comme test pour la PNH due à des mutations acquises du gène PIGA. Le test PNH peut révéler des anomalies dans d’autres déficiences d’ancrage GPI, mais le diagnostic repose principalement sur l’analyse moléculaire.
Analyse moléculaire
Le rendement diagnostique le plus élevé pour la GDC est le panel de séquençage génétique basé sur la prochaine génération ou le séquençage de l’exome clinique (CES). Le séquençage génétique relit la séquence nucléotidique, ou l’orthographe « lettre », des gènes pour déterminer s’il existe une modification qui affecte la fonction du gène. Le génome humain est constitué de 3 millions de nucléotides, mais seuls 1 à 2 % d’entre eux, appelés exons, sont traduits en un produit protéique fonctionnel. L’ADN non codant restant, intercalé entre les exons, qui n’est pas traduit, est appelé intron (74). La CES examine presque tous les exons connus des quelque 20 000 gènes du génome humain, qui représentent une minorité du matériel génétique des chromosomes mais sont les plus susceptibles de contenir des variantes pathogènes. Le CES peut également inclure le séquençage de l’ADN mitochondrial (ADNmt), qui interroge le petit ADN circulaire extranucléaire situé dans la mitochondrie et qui est exclusivement hérité par la mère.
Les résultats possibles du CES comprennent les résultats positifs, négatifs et les variants de signification inconnue. Un résultat positif signifie que des variants connus causant la maladie (c’est-à-dire pathogènes) sont identifiés, après quoi le diagnostic, l’histoire naturelle, le pronostic, le risque de récidive et les options de traitement peuvent être discutés. Un résultat négatif signifie qu’aucun variant pathogène détectable n’a été identifié. Un résultat négatif signifie qu’aucun variant pathogène détectable n’a été identifié. Un résultat négatif signifie qu’aucune variante pathogène détectable n’a été identifiée. Il faut s’attendre à des variations dans l’ADN de chaque individu, c’est pourquoi l’analyse simultanée d’échantillons parentaux à des fins de comparaison peut faciliter l’interprétation des résultats en laboratoire et en clinique. Le rendement diagnostique de la CES est estimé à 30-35 % et augmente avec le temps, à mesure que la découverte de gènes et les connaissances sur le génome humain continuent de progresser (75-77). La CES est de plus en plus souvent ordonnée comme le test génétique large de première ligne de choix en raison de son délai d’exécution rapide et de son faible coût relatif pour la quantité d’informations génétiques analysées. Les limites de la CES comprennent l’absence d’une sensibilité de 100 %, l’incapacité à détecter certains types de changements génétiques (par exemple, les délétions, les duplications, les répétitions trinucléotidiques, les mutations introniques profondes ou les défauts de méthylation), et le fait qu’un diagnostic peut ne pas fournir d’informations supplémentaires sur la maladie ou modifier la gestion.
Dans le rapport de la CES, les variantes pathogènes détectées par hasard dans les gènes associés à des conditions génétiques bien connues peuvent être signalées comme des résultats secondaires (78). Cette liste de maladies recommandées est établie par l’American College of Medical Genetics (ACMG). La loi sur la non-discrimination en matière d’information génétique (GINA) est un élément important à prendre en compte lorsqu’il s’agit de décider si l’on accepte ou non d’apprendre les découvertes fortuites (79). La GINA protège les individus contre l’utilisation abusive des informations génétiques dans le cadre de l’assurance maladie et de l’emploi, mais pas de l’assurance vie. La GINA protège les informations génétiques suivantes : les antécédents médicaux familiaux, les tests de porteurs, les tests génétiques prénataux, les tests de susceptibilité et les tests prédictifs, et l’analyse des tumeurs ou d’autres évaluations de gènes, de mutations ou de changements chromosomiques.
Gestion
La gestion de la GDC dépend largement des symptômes spécifiques de l’individu. Les symptômes récurrents chez les patients atteints de GDC comprennent un retard de croissance, un retard global de développement, des vomissements, des épisodes de type AVC et des anomalies squelettiques. Une coagulopathie clinique ou subclinique, une endocrinopathie, une hépatopathie et des anomalies cardiaques sont également fréquemment observées. Il est recommandé de procéder à des tests de laboratoire de base pour établir l’étendue de la maladie et d’effectuer une surveillance de routine, en particulier pour la PMM2-CDG. Il s’agit notamment des tests de la fonction hépatique, de l’albumine sérique, des tests de la fonction thyroïdienne, y compris la T4 libre, de la protéine C, de la protéine S, de l’antithrombine III, du facteur IX, de l’analyse d’urine et des gonadotrophines sériques et de l’hormone de croissance.
L’imagerie recommandée comprend l’échocardiogramme, l’échographie rénale, l’âge osseux, l’examen ophtalmologique pour l’évaluation du cristallin, de la rétine, de la mobilité oculaire et de la pression intraoculaire. Sauf indication contraire, les vaccinations de routine sont recommandées pour les adultes et les enfants atteints de GDC. Les titres d’anticorps doivent être obtenus après la vaccination car les patients peuvent avoir une réponse immunogène sous-optimale. Une réplétion prophylactique des facteurs de coagulation avant toute intervention chirurgicale peut être nécessaire si des carences existent au départ.
Une évaluation de génétique clinique doit être entreprise pour discuter des aspects héréditaires de la GDC, ainsi que pour établir un foyer médical pour ces patients complexes. Le foyer médical est généralement le service de génétique biochimique, bien que les départements de génétique, de neurodéveloppement ou de neurologie aient également joué ce rôle si un service de génétique biochimique dédié n’est pas disponible. L’orientation vers des spécialistes en gastroentérologie, en hématologie, en endocrinologie, en soutien nutritionnel, en orthophonie, en ergothérapie, en physiothérapie et en thérapie de l’alimentation, en orthopédie et en médecine de réadaptation est souvent nécessaire.
Thérapies ciblées et pronostic
Le traitement de la majorité des types de CDG est largement de soutien, à quelques exceptions près. Le MPI-CDG est le plus efficacement traitable de tous les CDG. Le mannose oral est converti en mannose-6-phosphate par les hexokinases intracellulaires, contournant ainsi le blocage enzymatique et produisant le substrat déficient. La supplémentation en mannose commence généralement à 1 g/kg de poids corporel par jour, réparti en 4 à 6 doses par jour. Alors que l’entéropathie à dégénérescence protéique, potentiellement mortelle, répond particulièrement bien au traitement par le mannose, la maladie hépatique dans le cas du MPI-CDG peut continuer à progresser. Les symptômes cliniques s’améliorent rapidement et les CDT de la transferrine se normalisent en quelques mois, bien que la maladie hépatique puisse continuer à progresser avec le traitement (45,80,81).
Il faut être prudent lors de la supplémentation en mannose pendant la grossesse, car l’administration de mannose chez des modèles de souris hypomorphes phosphomannose isomérase enceintes a entraîné une létalité embryonnaire et une cécité chez leurs petits (82). En outre, le mannose intraveineux a été associé à une diminution de la conscience et à des crises, qui se sont résolues avec l’administration de glucose (83).
Le traitement de la PMM2-CDG est largement de soutien et basé sur la symptomatologie. Cependant, de prochains essais cliniques sur la thérapie de remplacement du substrat du mannose-1-phosphate sont en cours de développement.
Pour les autres CDG, divers sucres simples oraux ont été étudiés dans le but d’améliorer théoriquement l’hypoglycosylation. Le fucose a été essayé pour le SLC35C1-CDG et le galactose pour le PGM1-CDG et le SLC35A2-CDG avec des résultats mitigés (84). Il a été démontré que le D-galactose à raison de 1,0-2,5 g/kg/jour (50 grammes maximum) améliore l’hypoglycémie, la coagulopathie et l’endocrinopathie chez les patients atteints de PGM1-CDG (85,86). Il a également été démontré que le galactose améliore l’endocrinopathie et la coagulopathie chez le TMEM165-CDG (87) et le SLC39A8-CDG. Une amélioration clinique considérable a également été rapportée chez des patients SLC39A8-CDG sous 15-20 mg/kg/jour de MnSO4 (88). Des essais cliniques sont en cours pour étudier l’utilité de la N-acétylmannosamine (ManNAc) dans le GNE-CDG (89), et plusieurs essais précliniques sont en cours pour d’autres CDG (90).
Malgré les progrès médicaux, une mortalité importante existe pour les enfants atteints de CDG au cours de la première année de vie en raison d’une défaillance multi-organique ou d’une infection grave (91). Les nourrissons atteints de GDC peuvent présenter une maladie multi-organique fulgurante, des convulsions intraitables ou une hypoalbuminémie sévère évoluant vers l’anasarque. Certains patients répondent à une diurèse agressive et au remplacement de l’albumine, tandis que d’autres sont réfractaires au traitement. Il a été démontré que le butyrate de sodium améliore le contrôle des crises dans les cas de CAD-CDG et de PIGM-CDG (92). Il a également été démontré que le régime cétogène diminue la fréquence des crises dans certains cas de PIGA-CDG (93). Pendant les épisodes de type AVC, l’hydratation par voie intraveineuse et le maintien d’une glycémie normale peuvent être utiles pendant que l’étiologie thrombotique ou hémorragique vasculaire sous-jacente est écartée.
Avec l’avènement des techniques d’édition du génome et une meilleure compréhension du mécanisme des maladies englobées dans l’ombrelle diagnostique du CDG, l’avenir du développement thérapeutique ciblé reste prometteur.
Remerciements
Nous tenons à remercier Lynne Wolfe, ARNP et Donna Krasnewich, MD, PhD pour nous avoir fourni des photos cliniques obtenues dans le cadre de l’étude Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). Nous tenons également à remercier Jenny Thies, MS, LGC pour son expertise en matière de conseil génétique.
Financement : IJ Chang est soutenu par les National Institutes of Health T32GM007454.
Note de bas de page
Conflits d’intérêts : Les auteurs n’ont aucun conflit d’intérêt à déclarer.
Consentement éclairé : Un consentement éclairé écrit a été obtenu des patients pour la publication de ce manuscrit et de toute image d’accompagnement.
- Jaeken J, Matthijs G. Troubles congénitaux de la glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation : a rapidly expanding disease family. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutations dans PMM2 qui causent des troubles congénitaux de la glycosylation, type Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation : an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. La prévalence de PMM2-CDG en Estonie basée sur les fréquences de porteurs de la population et les patients diagnostiqués. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Absence d’équilibre de Hardy-Weinberg pour la mutation PMM2 la plus prévalente dans les CDG-Ia (troubles congénitaux de la glycosylation de type Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sérum déficient en acide sialique et transferrine du liquide céphalorachidien dans un syndrome génétique nouvellement reconnu. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. Nomenclature GDC : il est temps de changer ! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders : fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Quoi de neuf dans la GDC ? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Troubles congénitaux de la glycosylation. Partie I. Défauts de la N-glycosylation des protéines. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Fonctions intracellulaires des glycanes N-liés. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Résultats cliniques, de laboratoire et moléculaires et données de suivi à long terme chez 96 patients français atteints de PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) et revue de la littérature. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. Une enquête nationale sur le PMM2-CDG en Italie : fréquence élevée d’une variante neurologique légère associée à la mutation L32R. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG en Afrique du Sud : Description génotype-phénotype de cinq nouveaux patients. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Différentes glycosyltransférases sont traitées de manière différentielle pour la sécrétion, la dimérisation et l’autoglycosylation. Glycobiologie 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In : Mora-Montes H. Éditeur. Glycanes : Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge : Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Troubles congénitaux de la glycosylation : autres causes d’ichtyose. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Manifestations cutanées dans la GDC. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. Le système IGF chez les enfants atteints de troubles congénitaux de la glycosylation. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. Un large spectre de présentations cliniques dans les troubles congénitaux de la glycosylation I : une série de 26 cas. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertal growth in congenital disorder of glycosylation type Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Caractéristiques cliniques et biochimiques du trouble congénital de la glycosylation de type Ic, le premier défaut reconnu du réticulum endoplasmique dans la synthèse des N-glycanes. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Congenital disorders of glycosylation : review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–une maladie métabolique héréditaire récemment découverte. Manifestations organiques multiples, incidence 1/80.000, difficile à traiter. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Ischémie myocardique transitoire grave causée par une cardiomyopathie hypertrophique chez un patient atteint d’un trouble congénital de la glycosylation de type Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Caractéristiques cliniques chez les adultes atteints de troubles congénitaux de la glycosylation de type Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Syndrome de la glycoprotéine déficiente en glucides. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Caractéristiques cliniques et moléculaires du trouble congénital de la glycosylation chez les patients présentant un modèle de sialotransferrine de type 1 et diverses origines ethniques. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. La modification O-GlcNAc de la protéine d’enveloppe du potyvirus Plum pox virus améliore l’infection virale. Virologie 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Patients scandinaves CDG-Ia : corrélation génotype/phénotype et origine géographique des mutations fondatrices. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 patients adultes français atteints de PMM2-trouble congénital de la glycosylation : résultat du phénotype pédiatrique classique et description d’un phénotype à apparition tardive. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Caractéristiques rétiniennes du trouble congénital de la glycosylation PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Trouble congénital de la glycosylation de type Ia (CDG-Ia) : spectre phénotypique du génotype R141H/F119L. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Fonction thyroïdienne chez les PMM2-CDG : approche diagnostique et proposition de prise en charge. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Trouble congénital de la glycosylation de type Ia présentant une ataxie cérébelleuse à début précoce chez un adulte. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a : trois frères et sœurs avec un phénotype neurologique léger. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Nouveaux troubles du métabolisme des glucides : troubles congénitaux de la glycosylation et leur impact sur le système endocrinien. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia : heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinologie du syndrome de la glycoprotéine déficiente en glucides de type 1 de la naissance à l’adolescence. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Fonction gonadique et hormones glycoprotéiques dans le syndrome de la glycoprotéine déficiente en glucides (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Le spectre clinique du déficit en phosphomannose isomérase, avec une évaluation du traitement au mannose pour le CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Complications cardiaques des troubles congénitaux de la glycosylation (CDG) : une revue systématique de la littérature. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Hydrops fetalis non immunisé récurrent associé au syndrome de la glycoprotéine déficiente en glucides. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency : a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Syndrome de la glycoprotéine déficiente en glucides de type Ib. Déficit en phosphomannose isomérase et thérapie au mannose. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Hypoglycémie sévère comme symptôme de présentation du syndrome de glycoprotéine déficiente en glucides. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency : Une nouvelle manifestation du syndrome des glycoprotéines déficientes en glucides pouvant être traitée par le mannose. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Carte génétique d’utilité clinique pour : ALG6 defective congenital disorder of glycosylation. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Mucus des voies respiratoires : De la production à la sécrétion. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Résultat clinique à long terme et variabilité phénotypique dans la calcinose tumorale familiale hyperphosphatémique et le syndrome d’hyperostose hyperphosphatémique causés par une nouvelle mutation GALNT3 ; rapport de cas et revue de la littérature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. L’héparane sulfate dans le développement, la croissance et la pathologie du squelette : le cas des exostoses multiples héréditaires. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency : homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Diverses affections musculo-squelettiques non inflammatoires. Calcinose tumorale familiale hyperphosphatémique (FGF23, GALNT3 et αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. L’ablation du gène Galnt3 entraîne de faibles concentrations de facteur de croissance des fibroblastes 23 (Fgf23) intactes dans la circulation et une hyperphosphatémie malgré une expression accrue du Fgf23. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Une mutation dans une enzyme de biosynthèse des gangliosides, ST3GAL5, entraîne le syndrome du poivre de sel &, un trouble neurocutané avec une altération de la glycosylation des glycolipides et des glycoprotéines. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Des mutations dans B4GALNT1 (GM2 synthase) sous-tendent un nouveau trouble de la biosynthèse des gangliosides. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. Les mutations PIGA provoquent des encéphalopathies épileptiques à début précoce et des caractéristiques distinctives. Neurologie. Neurologie 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Le syndrome des anomalies congénitales multiples-hypotonie-crises est causé par une mutation de PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. La mutation hypomorphe du promoteur de PIGM provoque une déficience héréditaire en glycosylphosphatidylinositol. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Des mutations hypomorphes de PGAP2, codant pour une protéine de remodelage des ancres GPI, provoquent une déficience intellectuelle autosomique récessive. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. Le phénotype d’une mutation germinale de PIGA : le gène muté somatiquement dans l’hémoglobinurie paroxystique nocturne. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Des mutations dans PIGO, un membre de la voie de synthèse des ancres GPI, provoquent une hyperphosphatasie avec retard mental. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. Un nouveau syndrome de déficience intellectuelle causé par une déficience de l’ancre GPI due à des mutations homozygotes dans PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Des mutations dans le gène du glycosylphosphatidylinositol PIGL causent le syndrome CHIME. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. Le spectre génotypique et phénotypique du déficit en PIGA. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. L’hémoglobinurie paroxystique nocturne (PNH) est causée par des mutations somatiques dans le gène PIG-A. EMBO J 1994;13:110-7.
- Brodsky RA. Hémoglobinurie paroxystique nocturne. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. L’impact de la spectrométrie de masse dans le diagnostic des troubles congénitaux de la glycosylation. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. La cardiomyopathie dilatée autosomique récessive due à des mutations DOLK résulte d’une O-mannosylation anormale du dystroglycane. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distributions des exons et des introns dans le génome humain. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Séquençage clinique de l’exome entier pour le diagnostic des troubles mendéliens. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. L’amélioration du rendement diagnostique par rapport aux panels de séquençage de gènes ciblés suggère un rôle pour le séquençage du génome entier comme test génétique de premier niveau. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. Le coût et le rendement diagnostique du séquençage de l’exome pour les enfants soupçonnés de troubles génétiques : une étude comparative. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommandations pour le signalement des résultats secondaires dans le séquençage clinique de l’exome et du génome, mise à jour 2016 (ACMG SF v2.0) : une déclaration de politique de l’American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Courants de l’éthique contemporaine. GINA, l’ADA et la discrimination génétique dans l’emploi. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Thrombo-embolie récurrente chez un enfant atteint d’un trouble congénital de la glycosylation (CDG) de type Ib et traitement au mannose. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Développement d’une maladie du foie malgré un traitement au mannose chez deux patients atteints de CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Les suppléments de mannose induisent la létalité embryonnaire et la cécité chez les souris hypomorphes à la phosphomannose isomérase. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Crises d’épilepsie et stupeur pendant un traitement au mannose par voie intraveineuse chez un patient atteint du syndrome CDG de type 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Supplémentation en glucose dans la déficience d’adhésion leucocytaire de type II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Déficience héréditaire en glycosylphosphatidyl inositol : un GDC traitable. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Supplémentation orale en D-galactose dans les cas de PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. La supplémentation en galactose chez les patients atteints de TMEM165-CDG sauve les défauts de glycosylation. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. Déficience en SLC39A8 : correction biochimique et amélioration clinique majeure par la thérapie au manganèse. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Thérapies orales à base de monosaccharides pour inverser l’hyposialylation rénale et musculaire dans un modèle murin de myopathie GNE. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. Thérapies GNE : From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Troubles de la glycosylation humaine. Marino PA, éditeur. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Thérapie ciblée pour la déficience héréditaire en GPI. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Régime cétogène – Un nouveau traitement pour l’encéphalopathie épileptique précoce due à un déficit en PIGA. Brain Dev 2016;38:848-51.