- Yleiskatsaus
- Epidemiologia
- Biokemiallinen luokittelu ja nimikkeistö
- Genetiikka
- Patofysiologia
- N-sidosteisten proteiinien glykosylaatiovirheet
- O-sidoksisen glykosylaation viat ja yhdistetyt N- ja O-sidoksisen glykosylaation viat
- Lipidiglykosylaatio ja GPI-ankkurin biosynteesin viat
- Kliiniset ilmenemismuodot
- N-sidosteisten proteiinien glykosylaatiohäiriöt
- PMM2-CDG (CDG-Ia, PMM2:n puutos)
- MPI-CDG (CDG-Ib, mannosefosfaatti-isomeraasin puutos)
- ALG6-CDG (glukosyylitransferaasi 1:n puutos)
- O-sidoksiset glykosylaatioviat ja yhdistetyt N- ja O-sidoksiset glykosylaatioviat
- Lipidiglykosylaation ja GPI-ankkurin biosynteesin puutokset
- Diagnostiikka
- N-sidoksissa olevien proteiinien glykosylaatiovirheet
- O-sidoksiset glykosylaatiovirheet ja yhdistetyt N- ja O-sidoksiset glykosylaatiovirheet
- Lipidiglykosylaatio- ja GPI-ankkurin biosynteesiviat
- Molekyylianalyysi
- Hoito
- Kohdennetut hoitomuodot ja ennuste
- Kiitokset
- Footnote
Yleiskatsaus
Glykosylaatio tarkoittaa sokerijäämien lisäämistä proteiineihin ja lipideihin erilaisissa solupoluissa. Synnynnäiset glykosylaatiohäiriöt (Congenital disorders of glycosylation, CDG) ovat geneettisesti ja kliinisesti heterogeeninen ryhmä, johon kuuluu yli sata sairautta, jotka johtuvat vioista glykaanisynteesin tai -modifikaatioreittien eri vaiheissa. Useimmat näistä monogeenisistä sairauksista periytyvät autosomaalisesti resessiivisesti, mutta myös autosomaalisesti dominoivia ja X-sidonnaisia muotoja on kuvattu.
CDG-taudit ilmenevät tyypillisesti moninaisina systeemisinä ilmenemismuotoina, joista yleisimpiä ovat kehitysviivästymä, menestymiskyvyttömyys, hypotonia, neurologiset poikkeavuudet, hepatopatia ja hyytymishäiriöt. Sairastuneilla henkilöillä voi esiintyä myös silmä-, iho- ja sydänsairauksia sekä kasvojen epämuodostumia. Vaikka neurologisia muutoksia ja kognitiivisia viivästymiä esiintyy suurimmalla osalla sairastuneista henkilöistä, on tiettyjä tapauksia ja jopa tyyppejä, joilla ei ole neurologisia oireita. Kun otetaan huomioon CDG:n laaja kliininen ja geneettinen etiologia, kliininen diagnoosi perustuu korkeaan epäilyindeksiin monisysteemisessä taudissa.
Serumin hiilihydraattipuutostransferriinianalyysi (CDT-analyysi) on ensilinjan seulontatesti potilailla, joilla epäillään CDG:tä, mutta sen havaitseminen rajoittuu N-glykosylaatiovirheisiin, joihin liittyy sialiinihappopuutoksia. Seuraavan linjan testeihin kuuluvat doliiniin sidoksissa olevien glykaanien analyysi ja geenitestit. Tämän eksponentiaalisesti kasvavien sairauksien ryhmän varhainen diagnosointi on tärkeää, sillä jotkin CDG:t ovat hoidettavissa. Glykosylaatiovirheiden hoito on pääasiassa tukihoitoa, vaikka MPI-CDG:lle, SLC35C1-CDG:lle, PIGM-CDG:lle ja PGM1-CDG:lle on saatavilla kohdennettuja hoitoja. Yksityiskohtaisia tietoja näistä hoidoista on jäljempänä kohdassa ”Kohdennetut hoidot ja ennuste”. Tässä katsauksessa keskitytään yleisimpiin CDG-tyyppeihin, joilla on tunnistettavat fenotyypit tai hoidot, ja kohderyhmänä ovat perusterveydenhuollon tarjoajat.
Epidemiologia
Kaikkityyppisten CDG:iden esiintyvyyttä ja yleisyyttä kokonaisuutena ei ole selvitetty hyvin, vaikka potilaita on raportoitu eri puolilla maailmaa melkein kaikilta etnisiltä taustoilta, ja molemmilla sukupuolilla on yhtä paljon sairauksia. Arvioitu esiintyvyys eurooppalaisessa ja afroamerikkalaisessa väestössä on 1/10 000 perustuen 53 geenin tunnettujen patogeenisten varianttien kantajataajuuksiin (1-4). Yleisimmin diagnosoidun CDG:n, PMM2-CDG:n, esiintyvyys on yksittäisten raporttien perusteella 1/20 000 hollantilaisessa väestössä ja 1/77 000 Virossa (5,6). Tähän mennessä useimmista CDG-tyypeistä on raportoitu alle 100 tapausta.
Biokemiallinen luokittelu ja nimikkeistö
Laaja-alaisesti CDG:t luokitellaan tällä hetkellä neljään luokkaan – (I) N-sidoksinen glykosylaatio, (II) O-sidoksinen glykosylaatio, (III) yhdistetty N- ja O-sidoksinen/moninkertainen glykosylaatio ja (IV) lipidien ja glykosyylifosfatidyyliinositolin (GPI) ankkurien biosynteesin häiriöt.
N-sidoksissa olevan proteiinin glykosylaatiovirhe PMM2-CDG, joka tunnettiin aiemmin nimellä CDG tyyppi Ia, oli ensimmäinen CDG, josta Jaeken raportoi vuonna 1980, ja se on edelleen ylivoimaisesti yleisin CDG tähän päivään mennessä (7). PMM2-CDG:tä kutsuttiin alun perin ”hiilihydraattipuutteisten glykoproteiinien oireyhtymäksi”, koska sairastuneilla henkilöillä havaittiin useita seerumin glykoproteiinipoikkeavuuksia seerumin transferriinin isoelektrisen fokusoinnin avulla. Aikaisemmin CDG:t luokiteltiin transferriinin isoformianalyysin mallien perusteella – tyypin I mallien katsottiin johtuvan doliiniin sidottujen glykaanien kokoonpano- ja siirtovirheistä, jotka paikallistuvat sytoplasmaan tai ER:ään, ja tyypin II mallien katsottiin johtuvan prosessointivirheistä Golgin laitteistossa. Tästä haarautumispisteestä CDG:t nimettiin sitten aakkosjärjestyksessä löytöjärjestyksessä.
Laajamittaisen molekyylidiagnostiikan yleistyttyä CDG-nimikkeistö päivitettiin vuonna 2008 täsmentämään sairauden molekulaarista etiologiaa, mikä heijastaa sellaisten polkujen ja häiriöiden eksponentiaalista kasvua, jotka eivät sopineet siististi aiemmin vakiintuneisiin kahtiajakautuneisiin luokkiin. Nykyisin CDG-nimikkeistö merkitään asianomaisen geenin nimellä (ei-kirjaimeton, geenien nimet osoitteessa www.genenames.org), jota seuraa -CDG (esim. PMM2-CDG) (8).
Genetiikka
Valtaosa synnynnäisistä glykosylaatiohäiriöistä periytyy autosomaalisen resessiivisen periytyvyyden mukaisesti, jolloin yksi mutaatio periytyy kummaltakin oireettomalta (kantajavanhemmalta). Geneettisen diagnoosin asettaminen edellyttää molekyylitestejä, yleensä seuraavan sukupolven sekvensointimenetelmillä. Vanhempien testaus tunnetun muunnoksen osalta voi vahvistaa periytymisen tai de novo -esiintymisen. Autosomaalisessa resessiivisessä periytymisessä sairastuneen yksilön sisarusten ja jokaisen raskauden uusiutumisriski on 25 % sairastuneelle, 50 % oireettomalle kantajalle ja 25 % sairastumattomalle.
Kourallinen CDG:tä on autosomaalista dominoivaa periytymistä (N-sidonnainen: GANAB-CDG, PRKCSH-CDG; O-sidoksissa: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Harvemmat ovat X-sidonnaisia (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). Useimmat CDG:n dominoivat ja jotkin X-sidoksissa olevat muodot johtuvat de novo -mutaatioista. Spesifiset sairaudet ja geenit on kuvattu jäljempänä kohdassa ”patofysiologia”.
Mutaatiotiedot kaikista julkaistuista CDG:n geeneistä ovat saatavilla Human Gene Mutation Database -tietokannassa (http://www.hgmd.cf.ac.uk/ac/index.php). Tietoa tietyistä geenimuunnoksista on saatavilla Leiden Open Variation Database -tietokannassa, johon on integroitu in silico -patogeenisuustyökaluja (http://www.lovd.nl/3.0/home). Kliinisiä yhteenvetoja tietyistä geeneistä löytyy Online Mendelian Inheritance in Man -verkkopalvelusta (http://www.omim.org/) tai suppeammassa laajuudessa osoitteesta GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Koska useimpien CDG:n alatyyppien kohdalla sairastuneiden potilaiden määrä on pieni, genotyypin ja fenotyypin välistä korrelaatiota on vaikea määrittää.
Patofysiologia
Tähän mennessä on raportoitu yli 130 CDG-tyyppiä (9,10). Koska glykosylaatioreitit ovat kaikkialla läsnä, CDG:t ovat biokemialliselta patogeneesiltään hyvin erilaisia. Lukuisat proteiinit ja lipidit (eli sfingolipidit ja glykolipidit) glykosyloituvat monosakkarideilla ja/tai oligosakkarideilla, joita kutsutaan yhdessä glykaaneiksi, eri solukompartimenteissa. Niiden subcellulaariset sijainnit ovat erilaisia, mutta useimmat viat esiintyvät ER:ssä tai Golgin laitteistossa. Yleisempien CDG:n kliiniset piirteet ja geneettinen etiologia poluittain on esitetty tiivistetysti taulukossa S1.
Proteiineista glykaanit kuvataan niiden kytkeytymisen perusteella polypeptidiketjuun N-glykaanit kiinnittyvät asparagiinin (Asn) amidiryhmään, kun taas O-glykaanit kiinnittyvät joko seriinin tai treoniinin hydroksyyliryhmään. N-glykaanisynteesi edellyttää nukleotidisidoksissa olevien sokerien vaiheittaista rakentamista sytosolissa, kokoamista endoplasmisessa retikulumissa ja prosessointia Golgin laitteessa. Sitä vastoin O-glykaanisynteesi edellyttää kokoonpanoa mutta ei prosessointia, joten O-glykosylaatiovirheitä esiintyy pääasiassa Golgin laitteessa.
N-sidosteisten proteiinien glykosylaatiovirheet
N-glykosylaatioon kuuluu hiilihydraattirakenteiden kovalenttinen kiinnittyminen Asn-jäännösten sivuketjujen amidiryhmään konsensuksen mukaisessa Asn-X-Ser/Thr-akseptorikohdassa, substraattipolypeptidin translokaatio endoplasmiseen retikulumiin uudelleenmuodostusta varten ja N-glykaaniketjun jatkomodifikaatio Golgin koneistossa (11,12). Vauriot missä tahansa synteesin, kokoonpanon ja prosessoinnin vaiheessa voivat johtaa kliiniseen sairauteen.
PMM2-CDG johtuu fosfomannomutaasi 2 (PMM2) -geenin patogeenisistä muunnoksista, jotka johtavat puutokseen PMM2-entsyymissä, joka katalysoi sytosolista mannoosi-6-fosfaatin muuntamista mannoosi-1-fosfaatiksi guanosiinidifosfaattimannoosisynteesin (GDP-mannoosisynteesin) toisessa vaiheessa. Useimmilla potilailla on yhdistetty heterotsygoottinen patogeeninen missense-mutaatio (www.lovd.nl/PMM2). Yleisin toistuva patogeeninen muunnos p.Arg141His esiintyy noin 40 prosentilla sairastuneista eurooppalaisista henkilöistä, ja myös p.Phe119Leu esiintyy usein Pohjois-Euroopassa (1). PMM2-CDG:n osalta on raportoitu genotyypin ja fenotyypin välisiä korrelaatioita (3,13,14).
MPI-CDG on autosomaalinen resessiivinen sairaus, jonka aiheuttavat patogeeniset variantit mannoosifosfaatti-isomeraasigeenissä (MPI), jotka johtavat fosfomannoosi-isomeraasin (MPI) puutteeseen. MPI katalysoi normaalisti GDP-mannoosisynteesin ensimmäistä vaihetta (eli fruktoosi-6-fosfaatin muuttamista mannoosi-6-fosfaatiksi), mutta fruktoosi-6-fosfaattia ei kerry solunsisäisesti, koska se voidaan metaboloida myös glykolyyttisen reitin kautta. Vaikka MPI-CDG on biokemiallisesti samankaltainen kuin PMM2-CDG, se ei näin ollen aiheuta yhtä merkittävää neurologista ja monisysteemistä haittaa. CDT on myös ensisijainen seulontatesti MPI-CDG:lle, joka osoittaa tyypin 1 mallia. Diagnoosi voidaan sitten vahvistaa molekyylitutkimuksella tai fibroblastien/leukosyyttien MPI-aktiivisuudella.
ALG6-CDG on resessiivinen sairaus, joka johtuu ALG6:n mutaatioista, jotka johtavat kolmen glukoosimolekyylin epänormaaliin kiinnittymiseen dolicholiin sidoksissa oleviin mannoosivälituotteisiin ja jälkikäteen tapahtuvaan seerumin glykoproteiinien hypoglykosylaatioon (15).
O-sidoksisen glykosylaation viat ja yhdistetyt N- ja O-sidoksisen glykosylaation viat
O-glykosylaatio käsittää hiilihydraattiketjujen vaiheittaisen lisäämisen proteiinien seriini-, treoniini- ja hydroksylyysiinijäämiin Golgi-laitteistossa sijaitsevien glykosyylitransferaasien toimesta (16). Useat erityyppiset O-sidoksiset glykaanit on yhdistetty ihmisen sairauksiin, ja ne on nimetty aminohappojäännökseen ensimmäisenä kiinnittyneen sokerin mukaan (17).
Lipidiglykosylaatio ja GPI-ankkurin biosynteesin viat
GPI-ankkurit ovat glykolipidejä, jotka kootaan peräkkäin endoplasmisessa retikuulumissa ja muokataan peräkkäin Golgissa. Entsyymipuutoksista johtuvat GPI-ankkurin biosynteesin häiriöt on nimetty aakkosjärjestyksessä löytöjärjestyksen mukaan eikä kronologisesti kokoonpanovaiheen mukaan. Kun GPI-ankkurit on syntetisoitu, ne sijaitsevat plasmakalvoilla ja sitovat satoja solupinnan proteiineja suorittaen lukuisia solutoimintoja. Useimmat näistä sairauksista ovat autosomaalisesti resessiivisesti periytyviä, merkittävänä poikkeuksena X-sidonnainen PIGA-puutos.
Kliiniset ilmenemismuodot
Glykosylaatioväylien ubiquitäärisen läsnäolon vuoksi käytännössä mikä tahansa elinjärjestelmä voi olla osallisena CDG:ssä, vaikkakin useimpiin tapauksiin liittyy neurologisia poikkeavuuksia. Joissakin CDG:ssä esiintyy iktyoosia, kuten MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (kuva 1) ja PIGL-CDG (18,19). Lähes kaikilla CDG:llä esiintyy monijärjestelmäistä sairautta ensimmäisten elinvuosien aikana, paitsi joillakin on vaikutusta vain yhteen elinjärjestelmään (esim, verkkokalvo DHDDS-CDG:ssä; hermo-lihasliitos ALG2-CDG:ssä, ALG14-CDG:ssä, CFPT1-CDG:ssä; aivot ST3GAL3-CDG:ssä, TUSC3-CDG:ssä; iho tai luurankolihas POGLUT1-CDG:ssä, POFUT1-CDG:ssä; rusto EXT1/EXT2-CDG:ssä; maksa TMEM199-CDG:ssä; punasolut SEC23B-CDG:ssä). Alkamisikä ja vaikeusaste voivat vaihdella vastasyntyneiden kuolemaan johtavasta vastasyntyneiden kuolemaan johtavasta lähes oireettomaan aikuisuuteen ja mihin tahansa siltä väliltä olevaan permutaatioon. Yleisimmin raportoituihin oirekokonaisuuksiin kuuluvat kehitysviivästymä, menestymishäiriö, hypotonia, neurologiset poikkeavuudet, hypoglykemia ja vaihtelevat maksa-, silmä-, iho-, ruoansulatuskanava-, immunologiset, luusto- ja hyytymispoikkeavuudet (19). Courtesy of NIH, Lynne Wolfe, CRNP, Donna Krasnewich, MD, PhD.
Monien CDG:n alatyyppien täydellinen fenotyyppi on vielä täysin määrittelemättä raportoitujen tapausten harvinaisuuden vuoksi. Siksi CDG:tä tulisi harkita missä tahansa monisysteemisen sairauden yhteydessä, erityisesti tapauksissa, joissa on neurologinen komponentti tai epäspesifinen kehitysviivästymä, jonka etiologia on epäselvä.
Vaikka monisysteemisten oireiden patofysiologiaa ei ole vielä selvitetty, tiettyjen glykosylaatioreittien ja erityisten kliinisten oireiden välistä suhdetta on selvitetty. Esimerkiksi monissa CDG-tyypeissä havaittu menestymättömyys johtuu useiden insuliinikasvureitin glykoproteiinien, kuten IGF-1:n, ALS:n ja IGFBP-3:n, hypoglykosylaatiosta ja heikentyneestä muodostumisesta (20). Kun ymmärrämme paremmin tätä monimutkaisten häiriöiden ryhmää, CDG tunnistetaan yhä useammin yksilöissä, joilla on vaikeasti selvitettävät diagnoosit. Taulukossa S1 on yhteenveto eri CDG:n elinjärjestelmien osallisuudesta. Seuraavassa käsittelemme CDG:n yleisimpien muotojen kliinisiä piirteitä ja muotoja, joilla on kohdennettuja hoitoja.
Täydellinen taulukko
N-sidosteisten proteiinien glykosylaatiohäiriöt
N-sidosteisten glykosylaatiohäiriöiden fenotyyppiä pidetään yleisimmin diagnosoitujen CDG:iden joukossa useasti klassisena esiintymismuotona. CDG:n fenotyyppinen kirjo on kuitenkin varsin moninainen, ja monissa CDG:ssä ei välttämättä esiinny PMM2-CDG:hen liittyviä stereotyyppisiä oireita.
PMM2-CDG (CDG-Ia, PMM2:n puutos)
PMM2-CDG on yleisin CDG:stä, ja maailmanlaajuisesti on raportoitu yli 700 tapausta. Sille on ominaista monisysteeminen vakava sairaus imeväisiässä, neurologinen sairaus ja kehitysviivästymä lapsuudessa ja/tai vakaa älyllinen kehitysvammaisuus aikuisuudessa (21,22).
Vauvaiässä PMM2-CDG:ssä esiintyy neurologisia poikkeavuuksia tyypillisesti pian syntymän jälkeen, nimittäin karsastusta ja epänormaaleja silmänliikkeitä, pikkuaivojen hypoplasiaa, hypotoniaa, psykomotorista hidastuneisuutta, ataksiaa, hypotoniaa ja hyporefleksiota. Lapsilla voi olla myös maksasairaus, nefroottinen oireyhtymä ja munuaiskystat, perikardiaalinen effuusio ja hypertrofinen kardiomyopatia, menestymishäiriö ja monielinvaurio, joka johtaa kuolemaan ensimmäisen elinvuoden aikana jopa 20 prosentilla sairastuneista (21,23-28).
Potilailla, joilla on PMM2-CDG:tä, on kuvattu dysmorfisten piirteiden muodostama yhdistelmä (kuvat 2,3). Näihin kuuluvat hypoplastinen pikkuaivo, kasvojen dysmorfiat (esim. suuret, dysplastiset korvat), käänteiset nännit ja rasvakudoksen epänormaali jakautuminen pakaroiden tai suprapubisen alueen päälle, mikä saattaa korjaantua iän myötä (14,21,29-32). Potilaiden on kuvattu olevan ulospäinsuuntautuneita ja iloisia. Esiintymismuoto on hyvin vaihteleva, vaikka karsastusta esiintyy yli 70 prosentilla sairastuneista potilaista (21,23,33-35). Käänteisiä nännejä ja epänormaaleja rasvapatjoja nähdään noin 25-50 %:lla potilaista (36).

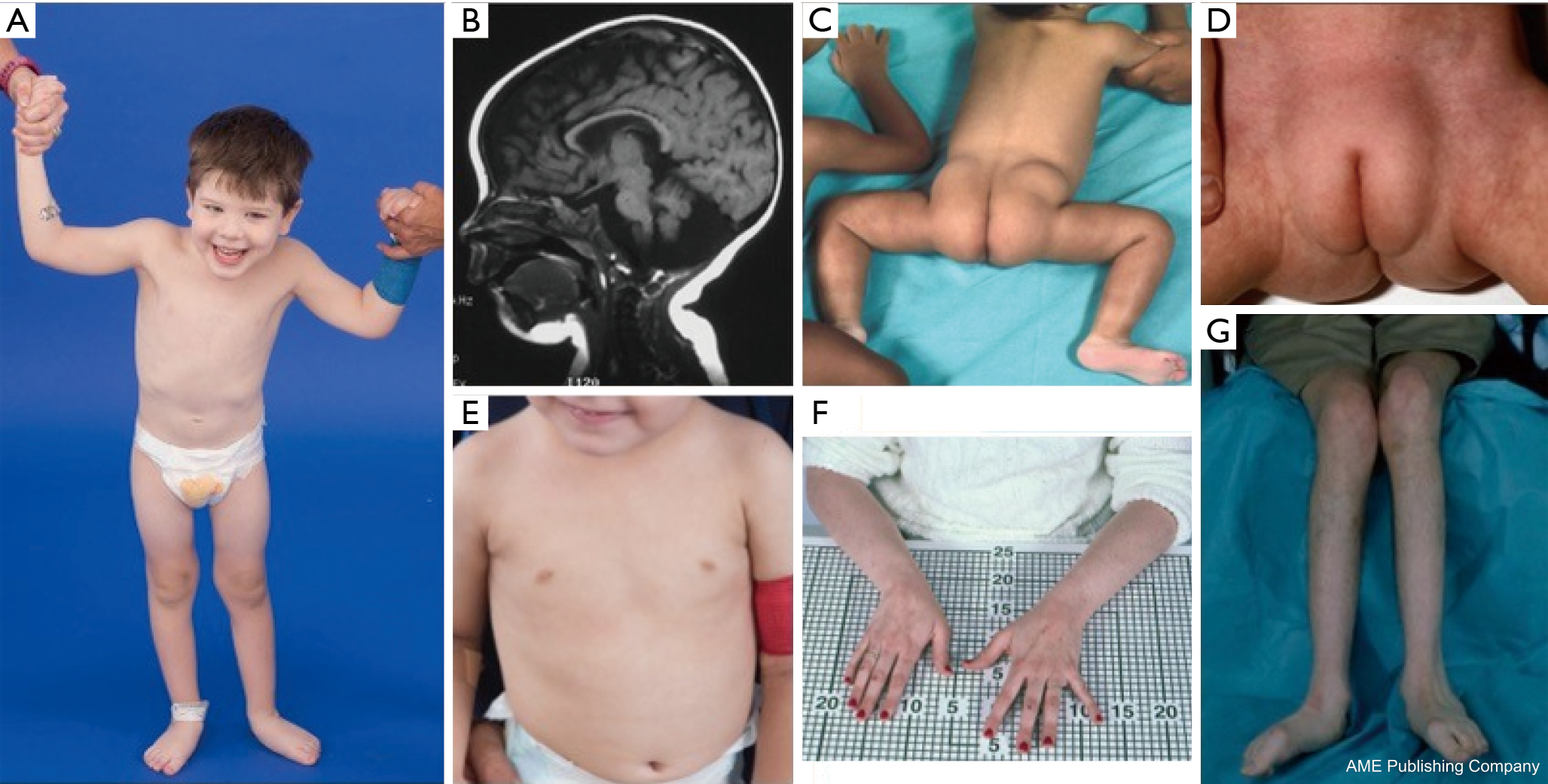
Lapsuudessa sairastuneille voi kehittyä retiniitti pigmentosa, aivohalvauksen kaltaisia kohtauksia ja kouristuskohtauksia, puheen ja motoriikan viivästymistä sekä perifeeristä neuropatiaa. Konstitutionaalisesti potilailla on yleisesti ruokinta- ja ruoansulatuskanavan poikkeavuuksista johtuva menestymishäiriö ja globaali kehitysviive. Maksan transaminaasit voivat olla koholla ilman kliinisiä seurauksia, ja yleensä ne normalisoituvat 5 vuoden ikään mennessä, mutta vaihtelevat toisinaan sairauden myötä (21,24). Maksabiopsiat ovat harvoin aiheellisia CDG:ssä, ellei epäillä maksan fibroosia (1). Kliininen kilpirauhasen vajaatoiminta on harvinaista, mutta CDG:tä sairastavilta potilailta tulisi mitata kilpirauhashormonit ja vapaa T4, jolloin voi ilmetä matalaa kilpirauhasen sitovaa globuliinia (TBG) ja ohimenevää kilpirauhasta stimuloivan hormonin (TSH) nousua (37). Maksa- ja sappiteiden epämuodostumia ei ole raportoitu PMM2-CDG-potilailla.
Aikuiset, joilla on PMM2-CDG, voivat elää 7. tai 8. vuosikymmenelle asti, ja heillä on vakaa kognitiivinen viive, perifeerinen neuropatia ja etenevä rintakehän ja selkärangan kyfoskolioosi, johon liittyy osteopenia tai osteoporoosi (34). Pikkuaivojen ataksia on yhä useammin tunnistettu oire yhdessä monisysteemisen osallistumisen kanssa (38-40). Endokriiniset poikkeavuudet, mukaan lukien hyperprolaktinemia, kasvuhormonin vapautuminen hyperglykemian yhteydessä, insuliiniresistenssi ja hyperinsulineminen hypoglykemia (41,42). Sairastuneilla naisilla hypogonadotrooppinen hypogonadismi voi johtaa sekundaarisen seksuaalisen kehityksen puuttumiseen tai munasarjojen puuttumiseen (41,43,44). Potilailla voi olla suurentunut tromboosiriski seerumin hyytymistekijöiden, kuten tekijöiden IV, IX ja XI, antitrombiini III:n, proteiini C:n ja proteiini S:n, pienentyneen määrän vuoksi (29).
MPI-CDG (CDG-Ib, mannosefosfaatti-isomeraasin puutos)
MPI-CDG on ainutlaatuinen, koska sairastuneilla potilailla on vain vähän tai ei lainkaan neurologisia oireita, ja jotkin taudin ilmenemismuodot ovat hoidettavissa suun kautta otettavalla annosteltavaksi tarkoitetulla annostelulla mannoosilla (2). Oireet ovat pääasiassa maksan ja suoliston välisiä ilman dysmorfisia piirteitä tai kognitiivisia viivästyksiä. Potilailla esiintyy tyypillisesti toistuvaa oksentelua, merkittävää hypoglykemiaa, kyvyttömyyttä menestyä, mahdollisesti henkeä uhkaavaa proteiinia tuhlaavaa enteropatiaa, maksan fibroottisia muutoksia ja sappiteiden laajentumia (45-51). Potilailla on suurentunut tromboottisten tapahtumien riski, koska seerumin proteiini C:n ja S:n pitoisuudet ja antitrombiini III:n pitoisuudet ovat alhaiset.
ALG6-CDG (glukosyylitransferaasi 1:n puutos)
ALG6-CDG on toiseksi yleisin N-glykosylaatiovirhe, jolle on ominaista samankaltainen, mutta lieväpiirteisempi fenotyyppi kuin PMM2-CDG. Potilailla, joilla on ALG6-CDG, on menestymishäiriöitä, kehitysviivästymiä, hypotoniaa, kouristuskohtauksia, karsastusta, ataksiaa, koagulopatiaa ja kasvojen dysmorfismia (esim. matalat korvat, hypertelorismi ja makroglossia). Samoin kuin MPI-CDG:llä, heillä voi olla myös proteiinia menettävää enteropatiaa. Lisäksi sairastuneilla potilailla voi olla luuston poikkeavuuksia, kuten brakydaktylia, sormien epämuodostumia ja skolioosia. Sairastuneilla potilailla ei tyypillisesti ole retinitis pigmentosaa tai pikkuaivojen hypoplasiaa (52).
O-sidoksiset glykosylaatioviat ja yhdistetyt N- ja O-sidoksiset glykosylaatioviat
Johtuen O-glykaanien huomattavasta esiintymisestä musiinia sisältävissä proteiineissa, mukaan lukien glykosaminoglykaanit (GAG:t), ja epiteelipinnoissa (53), GAG-synteesin häiriöt johtavat tyypillisesti luustohäiriöihin tai sidekudossairauksiin. Sairastuneilla potilailla voi esiintyä neurologisten oireiden lisäksi tuki- ja liikuntaelimistön, ihon ja nivelten poikkeavuuksia (esim. nivelten löysyys, multippelit eksostoosit, kondro/osteosarkoomat) (54-56). Esimerkiksi N-asetyyligalaktosaminyylitransferaasi 3 (GALNT3) O-glykosyloi fosfatuurihormoni FGF23:n, mikä estää proteolyyttisen pilkkoutumisen ja mahdollistaa sen ehjän erityksen. GALNT3-puutos johtaa familiaaliseen tumakalsinoosiin, jolle on ominaista hyperfosfatemia ja ektooppiset kalkkikertymät (57,58).
Lipidiglykosylaation ja GPI-ankkurin biosynteesin puutokset
Glykosfingolipidit ja niiden sialyloidut johdannaiset, gangliosidit, ekspressoituvat pääasiassa neuroneissa. Gangliosidien hajoamisen viat johtavat kertymiseen ja hyvin luonnehdittuihin lysosomaalisiin varastosairauksiin. Gangliosidien biosynteesin viat, kuten ST3GAL5-CDG ja B4GALNT1-CDG, ovat puolestaan erittäin harvinaisia ja johtavat vakaviin neurodegeneratiivisiin sairauksiin. Potilailla voi esiintyä spastista paraplegiaa, vakavaa älyllistä viivästymistä, epilepsiaa ja muita kuin neurologisia oireita, kuten luuston dysplasiaa, dysmorfisia piirteitä ja epänormaalia ihon pigmentaatiota (59,60).
Mutaatiot monissa GPI-ankkurin biosynteesireitillä olevissa geeneissä aiheuttavat monenlaisia moninaisia synnynnäisiä poikkeavuuksia, älyllistä vammaisuutta ja epilepsiaa. Parhaiten luonnehdittu GPI-biosynteesivirhe, X-ketjuinen PIGA-puutos, ilmenee lapsikouristuksina, joihin liittyy hypsarytmiaa, hypotoniaa, useita aivojen poikkeavuuksia ja kasvojen dysmorfismia. Potilailla voi olla myös vaihtelevia iho-, maksa-, sydän- ja munuaissairauksia (61-69). Jotkin PIGA:n mutaatiot aiheuttavat fenotyyppisesti erillisen sairauden, paroksysmaalisen yöllisen hemoglobinurian (PNH), joka on hankittu häiriö, jossa luuytimen toiminta pettää (70,71).
Diagnostiikka
Kun CDG:tä epäillään kliinisesti, ensimmäisenä askeleena tilataan biokemiallinen CDG-testi plasmasta tai seerumista, mukaan lukien CDT- ja N- glykaanitesti. Seerumin CDT- ja N-glykaanianalyysillä voidaan havaita vain N-glykosylaatiovirheet, joten niistä ei ole hyötyä eristettyjen O-glykosylaatio- tai GPI-ankkurivirheiden erottamisessa. Transferriinin isoformianalyysi tehtiin alun perin transferriinin isoelektrisen fokusoinnin avulla, koska N-glykaanisynteesin epäonnistuminen aiheuttaa sialiinihapon osittaisen puutteen, mikä muuttaa seerumin transferriinin varausta ja sen jälkeen sen katodista siirtymistä elektroforeettisella kentällä. Massaspektrometriaan perustuva transferriinin ja N-glykaanin analyysi on kuitenkin nyt suurelta osin korvannut isoelektrisen fokusoinnin tunnistamalla oligosakkaridien spesifiset muutokset massan ja varauksen perusteella (72).
N-sidoksissa olevien proteiinien glykosylaatiovirheet
Serumin transferriinin CDT-tulokset ilmoitetaan mono-oligosakkaridien/di-oligosakkaridien transferriinin suhteena, a-oligosakkaridien/di-oligosakkariditransferriinin, tri-sialo-/di-oligosakkariditransferriinin, apolipoproteiini CIII-1:n/apolipoproteiini CIII-2:n ja apolipoproteiini CIII-0:n/apolipoproteiini CIII-2:n suhde. Näihin kvantitatiivisiin tuloksiin liittyy myös tulkinta löydösten kuvioinnista.
Tyypin I kuvion transferriinin CDT:lle on ominaista lisääntyneet di- ja asialotransferriinikaistat, ja se viittaa puutteisiin N-glykaanisynteesissä sytosolissa tai endoplasmisessa retikulumissa. Tyypin II kuviolle on ominaista lisääntyneet di- ja asialotransferriinikaistaleet sekä tri- ja/tai monosialotransferriinikaistaleet, ja se viittaa vikoihin N-glykaanin prosessoinnissa Golgin laitteistossa (73).
Jos seerumin transferriinin CDT-tyypin tyyppi I -kuvio havaitaan, erotusdiagnoosien kärjessä tulisi olla PMM2-puutos tai MPI-puutos, sillä PMM2-CDG on yleisin CDG, ja MPI-CDG on hoidettavissa ja hoitamattomana se on mahdollisesti kuolemaan johtava. Diagnoosien erottamiseksi toisistaan olisi tehtävä N-glykaaniprofilointi, molekyylisekvensointi tai entsymaattinen testaus. PMM2-CDG:n tai MPI-CDG:n diagnoosi vahvistetaan molekyylitesteillä, joissa osoitetaan PMM2:n tai MPI:n kaksoispatogeeniset patogeeniset variantit, minkä jälkeen tutkitaan PMM- tai MPI-entsyymiaktiivisuus leukosyyteissä tai fibroblasteissa, jos geneettisten varianttien patogeenisyys on epävarma. N-glykaanianalyysi tai molekyylianalyysi erottaisi suurimman osan ALG-CDG:stä PMM2- tai MPI-CDG:stä (15).
Tyypin II seerumin transferriinin CDT-kuvio viittaa Golgin vikoihin, kuten N-asetyyliglukosaminiittitransferaasi (GnT) II:n puutokseen (CDG tyyppi IIA, MGAT2-CDG). Apolipoproteiini CIII:n (Apo-CIII) isomuodon analyysi on täydentävä testi tyypin II CDT-profiilille, sillä se mittaa muskiinityypin O-glykosylaatiovirheitä Golgin laitteistossa. CDT:n tai Apo-CIII:n herkkyys tyypin II CDG:n havaitsemisessa on rajallinen. Näin ollen N- ja O-glykaaniprofiilit ja molekyylipaneeli tai eksomisekvensointi olisi tehtävä, kun nämä kliiniset testit ovat käytettävissä. Transferriiniglykosylaatiomallit voivat normalisoitua satunnaisesti, joten toistuvat testit voivat olla aiheellisia potilaille, joilla on korkea epäilyindeksi. Vääriä positiivisia tuloksia voidaan saada potilailla, joilla on perinnöllisen fruktoosi-intoleranssin akuutti kriisi, galaktosemia, akuutti maksasairaus ja jotkin bakteeri-infektiot. Millään biokemiallisella CDG-testillä ei voida seuloa kaikkia CDG:tä, joten vaikka seulontatulokset olisivat normaalit, molekyyligeenipaneelitestaus tai eksomisekvensointi voidaan tehdä, jos on vahva kliininen epäilys. Sitä vastoin molekyyligeneettisten löydösten biokemiallinen ja toiminnallinen vahvistus on myös välttämätöntä, sillä suurimmalla osalla CDG-potilaista on ainakin yksi lievä ja usein uusi missense-mutaatio.
O-sidoksiset glykosylaatiovirheet ja yhdistetyt N- ja O-sidoksiset glykosylaatiovirheet
Diagnostiikka perustuu molekyylisen sekvensoinnin suorittamiseen, sillä transferriinin isomuoto-analyysi ei havaitsisi isolaarisesti esiintyviä O- glykosylaatiovikoja. Yhdistetyt N- ja O-sidoksiset glykosylaatioviat voidaan havaita CDT:llä, ApoCIII-analyysillä ja plasman N- ja O-glykaanianalyyseillä.
Lipidiglykosylaatio- ja GPI-ankkurin biosynteesiviat
Veren granulosyyttien virtaussytometrialla mitataan GPI-ankkuroitujen valkuaisaineiden, kuten CD16:n ja CD24:n, solujen pinnan ekspressiota. Valkosolujen tai punasolujen virtaussytometrinen analyysi tiettyjen GPI-ankkuroituneiden solupintaproteiinien osalta on saatavilla kliinisesti testinä PNH:n toteamiseksi, joka johtuu PIGA-geenin hankituista mutaatioista. PNH-testi voi paljastaa poikkeavuuksia muissa GPI-ankkuripuutoksissa, mutta diagnoosi perustuu useimmiten molekyylianalyysiin.
Molekyylianalyysi
Diagnostiikan korkein saanto CDG:n osalta on seuraavan sukupolven geenisekvensointipaneeliin perustuva seuraavan sukupolven geenisekvensointipaneeli tai kliininen eksomisekvensointi (CES). Geenisekvensoinnissa tarkistetaan geenien nukleotidisekvenssi eli ”kirjainkirjoitus” sen määrittämiseksi, onko geenin toimintaan vaikuttava muutos tapahtunut. Ihmisen perimässä on 3 miljoonaa nukleotidia, mutta vain 1-2 prosenttia näistä, ns. eksonit, käännetään toimivaksi proteiinituotteeksi. Jäljelle jäävää koodaamatonta DNA:ta, joka sijaitsee eksonien välissä ja jota ei käännetä, kutsutaan introneiksi (74). CES tutkii lähes kaikki tunnetut eksonit ihmisen genomin noin 20 000 geenistä, jotka muodostavat vähemmistön kromosomien geneettisestä materiaalista, mutta jotka todennäköisimmin sisältävät sairauksia aiheuttavia (patogeenisiä) variantteja. CES voi sisältää myös mitokondriaalisen DNA:n (mtDNA) sekvensoinnin, jossa tutkitaan mitokondrioissa sijaitsevaa pientä, ytimen ulkopuolista, pyöreää DNA:ta, joka periytyy yksinomaan äidiltä.
CES:n mahdollisia tuloksia ovat positiiviset, negatiiviset ja merkitykseltään tuntemattomat variantit. Positiivinen tulos tarkoittaa, että tunnetut tautia aiheuttavat (eli patogeeniset) variantit tunnistetaan, minkä jälkeen voidaan keskustella diagnoosista, luonnollisesta kulusta, ennusteesta, uusiutumisriskistä ja hoitovaihtoehdoista. Negatiivinen tulos tarkoittaa, ettei havaittavia patogeenisiä variantteja ole tunnistettu. Merkitykseltään tuntemattomat variantit (Variants of Unknown Significance, VUS) tarkoittaa, että vaikka geneettisiä muutoksia tunnistettiin, yksittäisestä geneettisestä muutoksesta ei ole riittävästi tietoa, jotta voitaisiin lopullisesti tietää, onko se tautia aiheuttava. Jokaisen yksilön DNA:ssa on odotettavissa eroja, joten vanhempien näytteiden samanaikainen testaus vertailua varten voi auttaa laboratorion ja kliinisen tutkimuksen tulosten tulkintaa. CES-testien diagnostinen saanto on arviolta 30-35 prosenttia, ja se kasvaa ajan mittaan, kun geenien löytäminen ja ihmisen genomia koskeva tietämys kehittyvät jatkuvasti (75-77). CES-testaus tilataan yhä useammin ensisijaiseksi laajaksi geneettiseksi testiksi, koska sen läpimenoaika on nopea ja suhteelliset kustannukset ovat alhaiset analysoituun geneettisen tiedon määrään nähden. CES:n rajoituksiin kuuluvat 100-prosenttisen herkkyyden puute, kyvyttömyys havaita tietyntyyppisiä geneettisiä muutoksia (esim. deleetioita, duplikaatioita, trinukleotidien toistoja, syviä intronisia mutaatioita tai metylaatiovirheitä) ja se, että diagnoosi ei välttämättä anna lisätietoa sairaudesta tai muuta hoitoa.
CES:n raportoinnissa voidaan raportoida toissijaisina löydöksinä sivutuotteina satunnaisesti havaittuja patogeenisiä variantteja geeneissä, jotka liittyvät tunnettuihin geneettisiin sairauksiin (78). Tämän suositeltujen sairauksien luettelon on kuratoinut American College of Medical Genetics (ACMG). Genetic Information Nondiscrimination Act (GINA) -laki on tärkeä näkökohta, kun päätetään, suostutaanko satunnaislöydösten selvittämiseen vai ei (79). GINA suojelee yksilöitä geneettisten tietojen väärinkäytöltä sairausvakuutuksessa ja työelämässä, mutta ei henkivakuutuksessa. GINA suojaa seuraavia geneettisiä tietoja: suvun sairaushistoria, kantajatestaus, synnytystä edeltävä geenitutkimus, alttius- ja ennustetestaus sekä kasvainten analyysi tai muu geenien, mutaatioiden tai kromosomimuutosten arviointi.
Hoito
CDG:n hoito riippuu pitkälti yksilön erityisistä oireista. Toistuvia oireita CDG:tä sairastavilla potilailla ovat menestymättömyys, globaali kehitysviive, oksentelu, aivohalvauksen kaltaiset kohtaukset ja luuston poikkeavuudet. Kliinistä tai subkliinistä koagulopatiaa, endokrinopatiaa, hepatopatiaa ja sydänvikoja esiintyy myös yleisesti. Peruslaboratoriokokeita taudin laajuuden määrittämiseksi ja rutiininomaista seurantaa suositellaan erityisesti PMM2-CDG:n osalta. Näihin kuuluvat maksan toimintakokeet, seerumin albumiini, kilpirauhasen toimintakokeet, mukaan lukien vapaa T4, proteiini C, proteiini S, antitrombiini III, tekijä IX, virtsa-analyysi sekä seerumin gonadotropiinit ja kasvuhormoni.
Suositeltuihin kuvantamistutkimuksiin kuuluvat kaikukardiografia, munuaisten ultraäänitutkimus, luuston ikä, silmätutkimus linssin arvioimiseksi, verkkokalvon, silmän liikkuvuuden ja silmänsisäisen silmänpaineen arviointi. Ellei toisin ilmoiteta, rutiinirokotuksia suositellaan CDG:hen sairastuneille aikuisille ja lapsille. Vasta-ainetitterit olisi määritettävä rokotuksen jälkeen, koska potilaiden immunogeeninen vaste voi olla suboptimaalinen. Hyytymistekijöiden profylaktinen täydentäminen ennen kirurgisia toimenpiteitä voi olla tarpeen, jos lähtötilanteessa on puutteita.
Kliinisen genetiikan arviointi olisi tehtävä CDG:n perinnöllisistä näkökohdista keskustelemiseksi sekä hoitokodin perustamiseksi näille monimutkaisille potilaille. Lääketieteellinen koti on tyypillisesti biokemiallisen genetiikan yksikkö, vaikka myös genetiikan, neurokehityksen tai neurologian osastot ovat toimineet tässä tehtävässä, jos erityistä biokemiallisen genetiikan yksikköä ei ole käytettävissä. Erikoislääkärin lähete gastroenterologian, hematologian, endokrinologian, ravitsemusterapian, puhe-, toiminta-, fysio- ja ruokintaterapian, ortopedian ja kuntoutuslääketieteen erikoislääkäreille on usein tarpeen.
Kohdennetut hoitomuodot ja ennuste
Vähemmistön CDG:n tyyppeistä hoito on pääosin tukihoitoa muutamaa poikkeusta lukuun ottamatta. MPI-CDG on kaikista CDG-tyypeistä tehokkaimmin hoidettavissa. Solunsisäiset heksokinaasit muuttavat oraalisen mannoosin mannoosi-6-fosfaatiksi, jolloin ohitetaan entsymaattinen esto ja tuotetaan puutteellinen substraatti. Mannoosilisäys aloitetaan yleensä 1 g/kg ruumiinpainoa päivässä jaettuna 4-6 annokseen päivässä. Mahdollisesti hengenvaarallinen proteiinia tuhlaava enteropatia reagoi erityisen hyvin mannoosihoitoon, mutta MPI-CDG:n maksasairaus voi edetä edelleen. Kliiniset oireet paranevat nopeasti ja transferriini CDT normalisoituu kuukausien kuluessa, vaikka maksasairaus voi edetä edelleen hoidon aikana (45,80,81).
Varovaisuutta on noudatettava annettaessa mannoosia lisäravinteena raskauden aikana, sillä mannoosin antaminen raskaana oleville hypomorfisille fosfomannoosi-isomeraasi-hiirimalleille johti alkion kuolettavuuteen ja pentujen sokeuteen (82). Lisäksi laskimonsisäiseen mannoosiin on liittynyt tajunnan heikkenemistä ja kouristuskohtauksia, jotka korjaantuivat glukoosin antamisen myötä (83).
PMM2-CDG:n hoito on suurelta osin tukihoitoa ja perustuu oireisiin. Tulevia kliinisiä tutkimuksia mannoosi-1-fosfaattisubstraatin korvaushoidosta on kuitenkin parhaillaan kehitteillä.
Muun CDG:n osalta on tutkittu erilaisia suun kautta otettavia yksinkertaisia sokereita, joiden tarkoituksena on teoriassa parantaa hypoglykosylaatiota. Fukoosia on kokeiltu SLC35C1-CDG:n osalta ja galaktoosia PGM1-CDG:n ja SLC35A2-CDG:n osalta vaihtelevin tuloksin (84). D-galaktoosin 1,0-2,5 g/kg/vrk (enintään 50 g) on osoitettu parantavan hypoglykemiaa, koagulopatiaa ja endokrinopatiaa PGM1-CDG:ssä (85,86). Galaktoosin on myös osoitettu parantavan endokrinopatiaa ja koagulopatiaa TMEM165-CDG:ssä (87) ja SLC39A8-CDG:ssä. Myös SLC39A8-CDG-potilailla on raportoitu huomattavaa kliinistä paranemista 15-20 mg/kg/vrk MnSO4:lla (88). Parhaillaan tehdään kliinisiä tutkimuksia N-asetyylimannosamiinin (ManNAc) käyttökelpoisuuden selvittämiseksi GNE-CDG:ssä (89), ja useita prekliinisiä tutkimuksia on käynnissä muiden CDG:iden osalta (90).
Lääketieteellisistä edistysaskeleista huolimatta CDG:tä sairastavilla lapsilla on merkittävä kuolleisuus ensimmäisen elinvuoden aikana monielinelinten vajaatoimintaan tai vakavaan infektioon (91). CDG:tä sairastavilla lapsilla voi esiintyä fulminantti monielinsairaus, vaikeasti hoidettavissa olevia kouristuksia tai vaikeaa hypoalbuminemiaa, joka etenee anasarkiaan. Jotkut potilaat reagoivat aggressiiviseen diureesiin ja albumiinikorvaukseen, kun taas toiset kieltäytyvät hoidosta. Natriumbutyraatin on osoitettu parantavan kohtausten hallintaa CAD-CDG:ssä ja PIGM-CDG:ssä (92). Ketogeenisen ruokavalion on myös osoitettu vähentävän kohtaustiheyttä joissakin PIGA-CDG-tapauksissa (93). Aivohalvauksen kaltaisten kohtausten aikana suonensisäisestä nesteytyksestä ja normaalin verensokerin ylläpitämisestä voi olla apua, kun taustalla oleva vaskulaarinen tromboottinen tai verenvuotoa aiheuttava etiologia suljetaan pois.
Genomin muokkaustekniikoiden yleistymisen ja CDG:n diagnostisen sateenvarjon piiriin kuuluvien tautien mekanismien paremman ymmärtämisen myötä kohdennetun terapeuttisen hoidon kehittämisen tulevaisuus pysyy lupaavana.
Kiitokset
Haluamme kiittää Lynne Wolfea, ARNP:tä ja Donna Krasnewichia, MD, PhD:tä siitä, että he antoivat meille kliiniset valokuvat, jotka saatiin osana Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789) -hanketta. Haluamme myös kiittää Jenny Thiesiä, MS, LGC hänen asiantuntemuksestaan geneettisessä neuvonnassa.
Rahoitus: IJ Changia tukee National Institutes of Health T32GM007454.
Footnote
Conflicts of Interest:
Tietoon perustuva suostumus:
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Hardy-Weinbergin tasapainon puuttuminen yleisimmän PMM2-mutaation osalta CDG-Ia:ssa (synnynnäiset glykosylaatiohäiriöt tyyppi Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialiinihappopuutteinen seerumin ja aivo-selkäydinnesteen transferriini äskettäin tunnistetussa geneettisessä oireyhtymässä. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG-nimikkeistö: muutoksen aika! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Congenital disorders of glycosylation. Osa I. Proteiinien N-glykosylaation viat. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. N-sidonnaisten glykaanien solunsisäiset toiminnot. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Kliiniset, laboratorio- ja molekyylilöydökset ja pitkäaikaisseurantatiedot 96 ranskalaiselta potilaalta, joilla on PMM2-CDG (fosfomannomutaasi 2:n synnynnäinen glykosylaatiohäiriö) ja kirjallisuuskatsaus. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG Etelä-Afrikassa: Viiden uuden potilaan genotyyppi-fenotyyppikuvaus. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Different glycosyltransferases are differentially processed for secretion, dimerization, and autoglycosylation. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Synnynnäiset glykosylaatiohäiriöt. In: Mora-Montes H. Editor. Glycans: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: Other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Skin manifestations in CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC ym. IGF-järjestelmä lapsilla, joilla on synnynnäisiä glykosylaatiohäiriöitä. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertaalinen kasvu synnynnäisessä glykosylaatiohäiriössä tyyppi Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Kliiniset ja biokemialliset ominaisuudet synnynnäisessä glykosylaatiohäiriössä tyyppi Ic, joka on ensimmäinen tunnistettu endoplasmisen retikulumin vika N-glykaanisynteesissä. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Synnynnäiset glykosylaatiohäiriöt: katsaus niiden molekulaarisiin perusteisiin, kliinisiin taudinkuviin ja erityishoitoihin. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–a recently discovered hereditary metabolic disease. Useita elimellisiä ilmenemismuotoja, esiintyvyys 1/80 000, vaikeasti hoidettavissa. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Hypertrofisen kardiomyopatian aiheuttama vakava ohimenevä sydänlihasiskemia potilaalla, jolla on synnynnäinen glykosylaatiohäiriö tyyppi Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Perikardiaalinen ja vatsanesteen kertyminen synnynnäisessä glykosylaatiotyypin Ia häiriössä. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Kliiniset piirteet aikuisilla, joilla on synnynnäinen glykosylaatiohäiriö tyyppi Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Hiilihydraattipuutteellinen glykoproteiinioireyhtymä. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, ym. synnynnäisen glykosylaatiohäiriön kliiniset ja molekulaariset piirteet potilailla, joilla on tyypin 1 sialotransferriinimalli ja erilainen etninen alkuperä. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modification of the coat protein of the potyvirus Plum pox virus enhances virus infection. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, ym. skandinaaviset CDG-Ia-potilaat: genotyypin ja fenotyypin korrelaatio ja perustajamutaatioiden maantieteellinen alkuperä. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 ranskalaista aikuispotilasta, joilla on PMM2- synnynnäinen glykosylaatiohäiriö: klassisen pediatrisen fenotyypin lopputulos ja myöhäisen fenotyypin kuvaaminen. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Synnynnäinen glykosylaatiohäiriö tyyppi Ia (CDG-Ia): R141H/F119L-genotyypin fenotyyppinen kirjo. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Thyroid function in PMM2-CDG: diagnostinen lähestymistapa ja ehdotettu hoito. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R ym. Synnynnäinen glykosylaatiohäiriö tyyppi 1a: kolme sisarusta, joilla lievä neurologinen fenotyyppi. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Hiilihydraattiaineenvaihdunnan uudet häiriöt: synnynnäiset glykosylaatiohäiriöt ja niiden vaikutus hormonitoimintaan. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogenity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Tyypin 1 hiilihydraattipuutteisen glykoproteiinioireyhtymän endokrinologia syntymästä nuoruuteen. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadien toiminta ja glykoproteiinihormonit hiilihydraattien puutteellisen glykoproteiinin (CDG) oireyhtymässä. Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Fosfomannoosi-isomeraasin puutoksen kliininen kirjo ja CDG-Ib:n mannoosihoidon arviointi. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Fosfomannoosi-isomeraasin puutos ja mannoosihoito. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sympt in phosphomannose isomerase deficiency: Uusi hiilihydraattipuutteisen glykoproteiinioireyhtymän ilmenemismuoto, jota voidaan hoitaa mannoosilla. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG6 defective congenital disorder of glycosylation. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: Tuotannosta eritykseen. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparaanisulfaatti luuston kehityksessä, kasvussa ja patologiassa: perinnöllinen multippeli eksostoosi. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homotsygoottinen p.R270C-mutaatio, johon liittyy founder-vaikutus, aiheuttaa Larsen of Reunion Island -oireyhtymän. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Miscellaneous non-inflammatory musculoskeletal conditions. Hyperfosfateeminen familiaalinen kasvainkalsinoosi (FGF23, GALNT3 ja αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Galnt3-geenin ablaatio johtaa alhaisiin verenkierrossa olevien ehjien fibroblastikasvutekijä 23:n (Fgf23) pitoisuuksiin ja hyperfosfatemiaan, vaikka Fgf23:n ilmentyminen on lisääntynyt. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Gangliosidien biosynteettisen entsyymin, ST3GAL5:n, mutaatio johtaa suola & pippurisyndroomaan, neurokutaaniseen häiriöön, johon liittyy muuttunut glykolipidien ja glykoproteiinien glykosylaatio. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA-mutaatiot aiheuttavat varhain alkavia epileptisiä enkefalopatioita ja erityispiirteitä. Neurology. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorfinen promoottorimutaatio PIGM:ssä aiheuttaa perinnöllisen glykosyylifosfatidylinositolipuutoksen. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorfiset mutaatiot GPI-ankkuria uudelleenmuokkaavaa proteiinia koodaavassa PGAP2:ssa aiheuttavat autosomaalisesti resessiivistä älyllistä kehitysvammaisuutta. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J 1994;13:110-7.
- Brodsky RA. Paroksismaalinen yöllinen hemoglobinuria. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. Massaspektrometrian vaikutus synnynnäisten glykosylaatiohäiriöiden diagnostiikassa. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomaalinen resessiivinen dilatoiva kardiomyopatia, joka johtuu DOLK-mutaatioista, johtuu poikkeavasta dystroglykaanin O-mannosylaatiosta. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Exonien ja intronien jakautuminen ihmisen genomissa. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Currents in contemporary ethics. GINA, ADA ja geneettinen syrjintä työelämässä. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Toistuva tromboembolia lapsella, jolla on synnynnäinen glykosylaatiohäiriö (CDG) tyyppi Ib ja hoito mannoosilla. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Maksasairauden kehittyminen mannoosihoidosta huolimatta kahdella CDG-Ib-potilaalla. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannoosilisät aiheuttavat alkion letaliteettia ja sokeutta fosfomannoosi-isomeraasihypomorfisilla hiirillä. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Kohtaukset ja stupor suonensisäisen mannoosihoidon aikana potilaalla, jolla on CDG-oireyhtymä tyyppi 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Fukoosilisäys leukosyyttien adheesiopuutostyypin II yhteydessä. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Perinnöllinen glykosyylifosfatidyyli-inositolin puutos: hoidettavissa oleva CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Oral D-galaktoosilisäys PGM1-CDG:ssä. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Galaktoosilisäys potilailla, joilla on TMEM165-CDG, pelastaa glykosylaatiovirheet. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8-puutos: biokemiallinen korjaus ja merkittävä kliininen paraneminen mangaanihoidolla. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Oral monosakkaridihoidot munuaisten ja lihasten hyposialylaation kumoamiseksi GNE-myopatian hiirimallissa. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Ihmisen glykosylaatiohäiriöt. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogeeninen ruokavalio – PIGA-puutoksesta johtuvan varhaisen epileptisen enkefalopatian uusi hoito. Brain Dev 2016;38:848-51.