- Přehled
- Epidemiologie
- Biochemická klasifikace a nomenklatura
- Genetika
- Patofyziologie
- N-vázané defekty glykosylace proteinů
- Defekty O-vázané glykosylace a kombinované defekty N- a O-vázané glykosylace
- Glykosylace lipidů a defekty biosyntézy GPI kotev
- Klinické projevy
- N-vázané defekty glykosylace proteinů
- PMM2-CDG (CDG-Ia, deficit PMM2)
- MPI-CDG (CDG-Ib, deficit mannosafosfát izomerázy)
- ALG6-CDG (deficit glukosyltransferázy 1)
- O-vázané defekty glykosylace a kombinované defekty N- a O-vázané glykosylace
- Defekty glykosylace lipidů a biosyntézy GPI kotev
- Diagnostika
- N-vázané defekty glykosylace proteinů
- O-vázané glykosylační defekty a kombinované N- a O-vázané glykosylační defekty
- Defekty glykosylace lipidů a biosyntézy GPI kotev
- Molekulární analýza
- Management
- Cílené terapie a prognóza
- Poděkování
- Poznámka pod čarou
Přehled
Glykosylace je proces přidávání cukerných zbytků k proteinům a lipidům v různých buněčných drahách. Vrozené poruchy glykosylace (CDG) jsou geneticky a klinicky heterogenní skupinou více než stovky onemocnění způsobených defekty v různých krocích na cestě syntézy nebo modifikace glykanů. Většina těchto monogenních onemocnění má autozomálně recesivní dědičnost, ale byly popsány také autozomálně dominantní formy a formy vázané na chromozom X.
CDG se obvykle projevují multisystémovými projevy, nejčastěji opožděním vývoje, neprospíváním, hypotonií, neurologickými abnormalitami, hepatopatií a koagulopatií. Postižení jedinci mohou mít také oční, kožní a srdeční onemocnění a obličejové dysmorfismy. Ačkoli neurologické změny a kognitivní opoždění jsou pozorovány u většiny postižených jedinců, existují určité případy a dokonce i typy, které neurologické projevy nemají. Vzhledem k široké klinické a genetické etiologii CDG se klinická diagnóza opírá o vysoký index podezření u multisystémového onemocnění.
Analýza karbohydrát deficientního transferinu (CDT) v séru je screeningovým testem první volby u pacientů s podezřením na CDG, ale je omezena v detekci na defekty N-glykosylace s nedostatkem kyseliny sialové. Testy další linie zahrnují analýzu glykanů vázaných na dolichol a genetické testování. Včasná diagnostika této skupiny exponenciálně rostoucích onemocnění je důležitá, protože některé CDG jsou léčitelné. Léčba glykosylačních defektů je převážně podpůrná, i když je k dispozici cílená léčba MPI-CDG, SLC35C1-CDG, PIGM-CDG a PGM1-CDG. Podrobnosti o této léčbě jsou uvedeny níže v části „Cílená léčba a prognóza“. V tomto přehledu se zaměříme na nejčastější typy CDG s rozpoznatelnými fenotypy nebo léčebnými postupy, přičemž cílovou skupinou jsou poskytovatelé primární péče.
Epidemiologie
Incidence a prevalence všech typů CDG v souhrnu nejsou přesně stanoveny, ačkoli celosvětově jsou hlášeni pacienti téměř všech etnik a obě pohlaví jsou postižena stejně. Odhadovaná prevalence v evropské a afroamerické populaci je 1/10 000 na základě frekvencí nosičství známých patogenních variant v 53 genech (1-4). Prevalence nejčastěji diagnostikované CDG, PMM2-CDG, se na základě ojedinělých zpráv pohybuje od 1/20 000 v nizozemské populaci a 1/77 000 v Estonsku (5,6). Dosud bylo u většiny typů CDG hlášeno méně než 100 případů.
Biochemická klasifikace a nomenklatura
V současnosti se CDG obecně dělí do čtyř kategorií – (I) N-vázaná glykosylace, (II) O-vázaná glykosylace, (III) kombinovaná N- a O-vázaná/vícečetná glykosylace a (IV) defekty biosyntézy lipidových a glykosylfosfatidylinositolových (GPI) kotev.
Defekt N-vázané glykosylace proteinů PMM2-CDG, dříve známý jako CDG typu Ia, byl prvním CDG, který popsal Jaeken v roce 1980, a dodnes zůstává zdaleka nejčastějším CDG (7). PMM2-CDG byl původně nazván „syndrom glykoproteinového deficitu karbohydrátů“ vzhledem k četným abnormalitám sérových glykoproteinů pozorovaným při izoelektrické fokusaci sérového transferinu u postižených jedinců. Historicky byly CDG klasifikovány podle vzorců analýzy izoforem transferinu – vzorce typu I byly přisuzovány defektům sestavování a přenosu glykanů vázaných na dolichol lokalizovaným v cytoplazmě nebo ER a vzorce typu II byly přisuzovány defektům zpracování v Golgiho aparátu. Od tohoto rozvětvení se pak CDG pojmenovávaly abecedně podle pořadí objevu.
S příchodem rozšířené molekulární diagnostiky byla nomenklatura CDG v roce 2008 aktualizována tak, aby specifikovala molekulární etiologii onemocnění, což odráželo exponenciální nárůst cest a poruch, které se do dříve zavedených dichotomických kategorií úhledně nevešly. V současné době se CDG nomenklatura označuje názvem postiženého genu (neitalizováno, názvy genů na www.genenames.org), za kterým následuje -CDG (např. PMM2-CDG) (8).
Genetika
Převážná většina vrozených poruch glykosylace se dědí autozomálně recesivním způsobem, přičemž každý z asymptomatických rodičů (přenašečů) zdědí jednu mutaci. Ke stanovení genetické diagnózy je nezbytné molekulární vyšetření, obvykle metodami sekvenování nové generace. Testování rodičů na známou variantu může potvrdit dědičnost oproti výskytu de novo. U autozomálně recesivní dědičnosti je riziko opakování u sourozenců a každého těhotenství postiženého jedince 25 % pro postižení, 50 % pro asymptomatické přenašeče a 25 % pro nepostižení.
Několik CDG má autozomálně dominantní dědičnost (N-vázanou: GANAB-CDG, PRKCSH-CDG; O-vázaná: (EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Méně je X-vázaných (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). Většina dominantních a některé X-vázané formy CDG jsou způsobeny mutacemi de novo. Konkrétní onemocnění a geny jsou popsány níže v části „patofyziologie“.
Údaje o mutacích všech publikovaných genů pro CDG jsou k dispozici v databázi Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Informace o konkrétních variantách genů jsou k dispozici na adrese Leiden Open Variation Database s integrovanými nástroji patogenity in silico (http://www.lovd.nl/3.0/home). Klinické přehledy pro konkrétní geny lze nalézt na adrese Online Mendelian Inheritance in Man (http://www.omim.org/) nebo v omezenějším rozsahu na adrese GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Vzhledem k malému počtu postižených pacientů u většiny podtypů CDG je obtížné stanovit korelaci mezi genotypem a fenotypem.
Patofyziologie
Dosud bylo popsáno více než 130 typů CDG (9,10). Vzhledem k všudypřítomnosti glykosylačních drah jsou CDG ve své biochemické patogenezi velmi rozmanité. Četné proteiny a lipidy (tj. sfingolipidy a glykolipidy) podléhají v různých buněčných kompartmentech glykosylaci monosacharidy a/nebo oligosacharidy, souhrnně označovanými jako glykany. Jejich subcelulární umístění je různé, ale většina defektů se vyskytuje v ER nebo Golgiho aparátu. Klinické příznaky a genetická etiologie častějších CDG podle cest jsou shrnuty v tabulce S1.
Mezi proteiny jsou glykany popsány podle jejich vazby na polypeptidový řetězec – N-glykany jsou navázány na amidovou skupinu asparaginu (Asn), zatímco O-glykany jsou navázány na hydroxylovou skupinu serinu nebo treoninu. Syntéza N-glykanů vyžaduje postupnou výstavbu nukleotidově vázaných cukrů v cytosolu, sestavení v endoplazmatickém retikulu a zpracování v Golgiho aparátu. Naproti tomu syntéza O-glykanů vyžaduje sestavení, ale žádné zpracování, proto se defekty O-glykosylace vyskytují převážně v Golgiho aparátu.
N-vázané defekty glykosylace proteinů
N-glykosylace zahrnuje kovalentní připojení sacharidových struktur k amidové skupině postranního řetězce Asn zbytků v rámci konsenzuálního akceptorového místa Asn-X-Ser/Thr, translokaci substrátového polypeptidu do endoplazmatického retikula k přestavbě a další modifikaci N-glykanového řetězce v Golgiho aparátu (11,12). Defekty kdekoli na cestě syntézy, montáže a zpracování mohou vést ke klinickému onemocnění.
PMM2-CDG je způsobena patogenními variantami v genu pro fosfomannomutázu 2 (PMM2), což vede k deficitu enzymu PMM2, který katalyzuje cytosolickou přeměnu manóza-6-fosfátu na manóza-1-fosfát ve druhém kroku syntézy manózy guanosindifosfátu (GDP). Většina pacientů má složené heterozygotní patogenní missense mutace (www.lovd.nl/PMM2). Nejčastěji se opakující patogenní varianta p.Arg141His se vyskytuje přibližně u 40 % postižených jedinců evropského původu a p.Phe119Leu se také často vyskytuje v severní Evropě (1). U PMM2-CDG byly popsány korelace mezi genotypem a fenotypem (3,13,14).
MPI-CDG je autozomálně recesivní porucha způsobená patogenními variantami v genu pro manózafosfát izomerázu (MPI), které vedou k deficienci fosfomanóza izomerázy (MPI). MPI normálně katalyzuje první krok syntézy GDP-manózy (tj. přeměnu fruktóza-6-fosfátu na manóza-6-fosfát), ale fruktóza-6-fosfát se intracelulárně nehromadí, protože může být metabolizován také glykolytickou cestou. Ačkoli je tedy MPI-CDG biochemicky podobný PMM2-CDG, nezpůsobuje tak významné neurologické a multisystémové postižení. CDT je také screeningovým testem volby pro MPI-CDG, který vykazuje vzorec typu 1. Diagnóza pak může být potvrzena molekulárně nebo pomocí aktivity MPI fibroblastů/leukocytů.
ALG6-CDG je recesivní onemocnění způsobené mutacemi v ALG6, které vedou k abnormálnímu navázání tří molekul glukózy na intermediáty manózy vázané na dolichol a následné hypoglykosylaci sérových glykoproteinů (15).
Defekty O-vázané glykosylace a kombinované defekty N- a O-vázané glykosylace
O-glykosylace zahrnuje postupné přidávání sacharidových řetězců k serinovým, treoninovým a hydroxylysinovým zbytkům proteinů glykosyltransferázami v Golgiho aparátu (16). S lidskými chorobami je spojeno několik typů O-vázaných glykanů, pojmenovaných podle prvního cukru připojeného ke zbytku aminokyseliny (17).
Glykosylace lipidů a defekty biosyntézy GPI kotev
GPI kotvy jsou glykolipidy, které procházejí postupným sestavováním v endoplazmatickém retikulu a modifikacemi v Golgiho aparátu. Poruchy biosyntézy kotev GPI způsobené nedostatkem enzymů jsou pojmenovány abecedně podle pořadí objevení, nikoliv chronologicky podle kroku sestavení. Po syntéze se kotvy GPI nacházejí na plazmatických membránách a vážou stovky proteinů na povrchu buněk, čímž plní množství buněčných funkcí. Většina těchto onemocnění je autozomálně recesivní s pozoruhodnou výjimkou X-vázaného deficitu PIGA.
Klinické projevy
Vzhledem k všudypřítomnosti glykosylačních drah může být u CDG postižen prakticky jakýkoli orgánový systém, ačkoli většina případů zahrnuje neurologické abnormality. Některé CDG se projevují ichtyózou, včetně MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (obrázek 1) a PIGL-CDG (18,19). Téměř všechny CDG se projevují multisystémovým onemocněním během několika prvních let života, s výjimkou některých, které postihují pouze jeden orgánový systém (např, sítnici u DHDDS-CDG; nervosvalové spojení u ALG2-CDG, ALG14-CDG, CFPT1-CDG; mozek u ST3GAL3-CDG, TUSC3-CDG; kůži nebo kosterní svalstvo u POGLUT1-CDG, POFUT1-CDG; chrupavku u EXT1/EXT2-CDG; játra u TMEM199-CDG; červené krvinky u SEC23B-CDG). Věk nástupu a závažnost se mohou pohybovat od novorozenecké letality až po téměř asymptomatickou dospělost a jakoukoli permutaci mezi nimi. Nejčastěji uváděná konstelace příznaků zahrnuje opoždění vývoje, neprospívání, hypotonii, neurologické abnormality, hypoglykémii a variabilní jaterní, oční, kožní, gastrointestinální, imunologické, kosterní a koagulační abnormality (19).
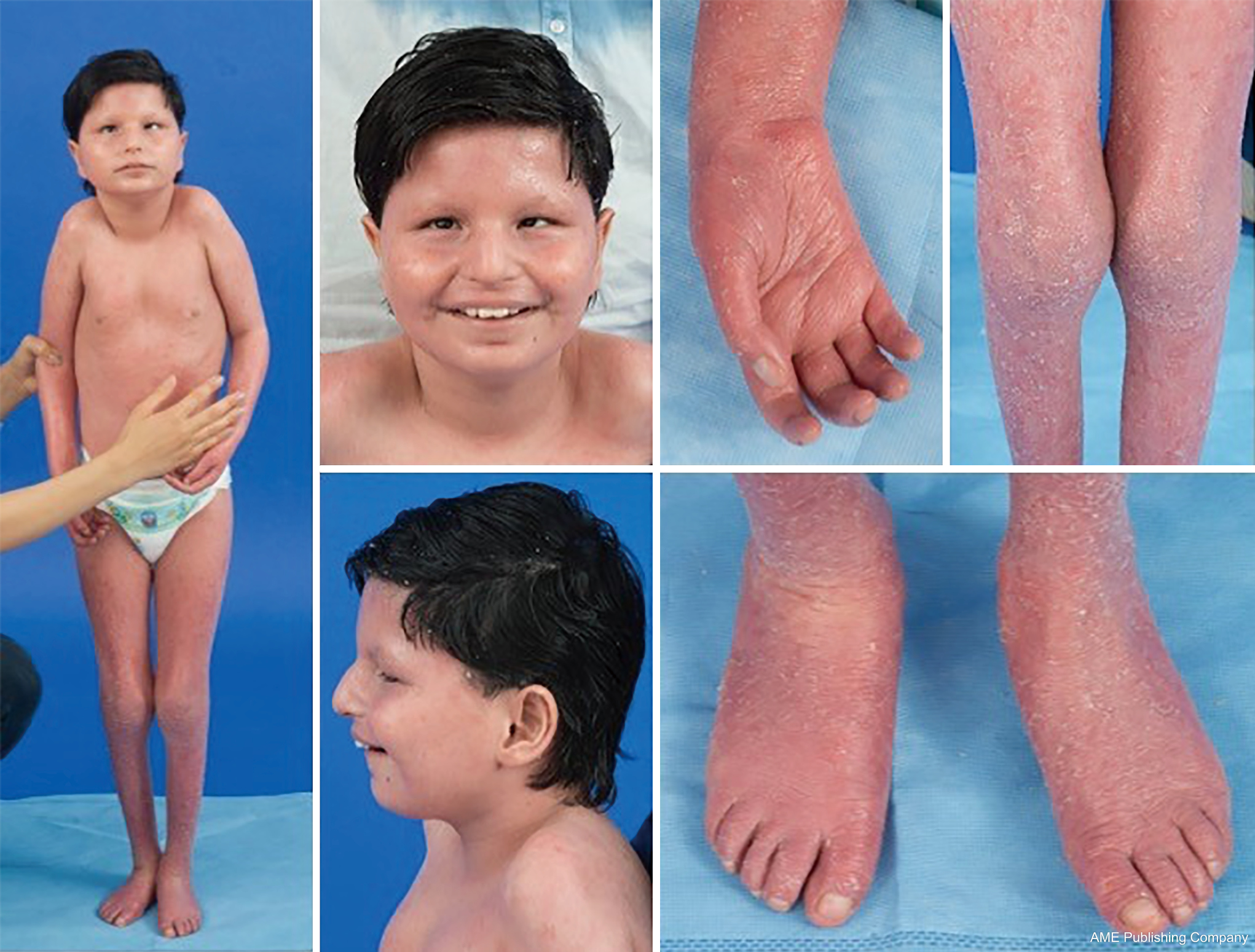
Kompletní fenotyp mnoha podtypů CDG není vzhledem k vzácnosti hlášených případů dosud plně vymezen. Proto by se na CDG mělo pomýšlet při jakémkoli multisystémovém onemocnění, zejména v případech s neurologickou složkou nebo nespecifickým vývojovým opožděním s nejasnou etiologií.
Ačkoli patofyziologie multisystémových příznaků stále není objasněna, byl objasněn vztah mezi určitými glykosylačními cestami a specifickými klinickými příznaky. Například neprospívání pozorované u mnoha typů CDG lze přičíst hypoglykosylaci a poruše tvorby několika glykoproteinů v rámci inzulinové růstové dráhy včetně IGF-1, ALS a IGFBP-3 (20). S tím, jak lépe rozumíme této skupině komplexních poruch, jsou CDG stále častěji rozpoznávány u jedinců s nepolapitelnými diagnózami. Postižení orgánových systémů u různých CDG je shrnuto v tabulce S1. Níže se budeme zabývat klinickými příznaky nejčastějších forem a forem s cílenou léčbou CDG.
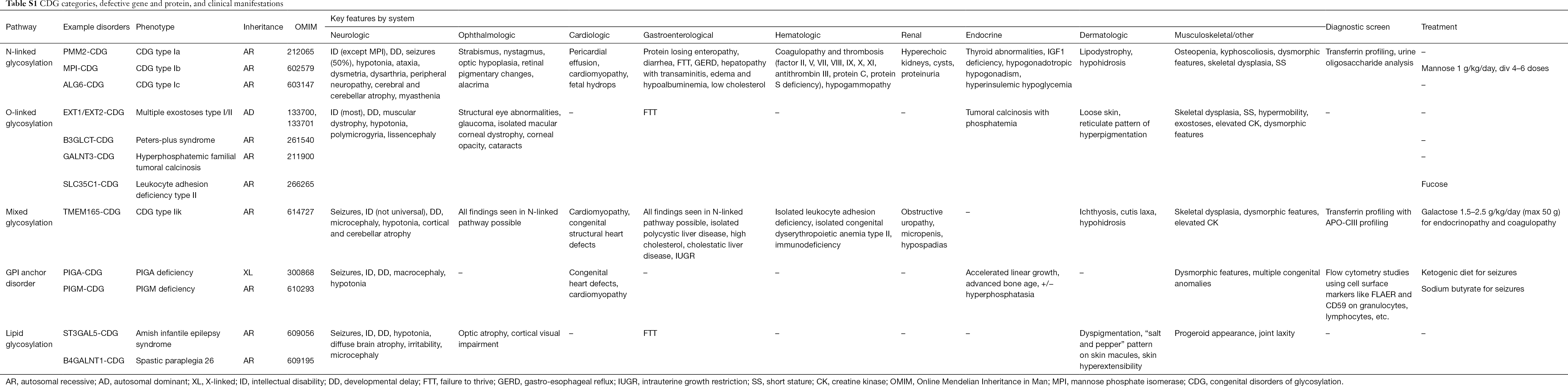
Úplná tabulka
N-vázané defekty glykosylace proteinů
Jako nejčastěji diagnostikovaná CDG je fenotyp N-vázaných poruch glykosylace často zvěstován jako klasický projev. Fenotypové spektrum CDG je však poměrně rozmanité a mnoho CDG se nemusí projevovat stereotypními příznaky spojenými s PMM2-CDG.
PMM2-CDG (CDG-Ia, deficit PMM2)
PMM2-CDG je nejčastější CDG, celosvětově je hlášeno více než 700 případů. Je charakterizována multisystémovým závažným onemocněním v kojeneckém věku, neurologickým onemocněním a opožděním vývoje v dětství a/nebo stabilním intelektuálním postižením v dospělosti (21,22).
V kojeneckém věku se PMM2-CDG projevuje neurologickými abnormalitami typicky krátce po narození, a to strabismem a abnormálními očními pohyby, hypoplazií mozečku, hypotonií, psychomotorickou retardací, ataxií, hypotonií a hyporeflexií. Kojenci mohou mít také jaterní onemocnění, nefrotický syndrom a ledvinové cysty, perikardiální výpotek a hypertrofickou kardiomyopatii, neprospívání a multiorgánové selhání, které až u 20 % postižených jedinců vede k úmrtí během prvního roku života (21,23-28).
U pacientů s PMM2-CDG byla popsána konstelace dysmorfických rysů (obrázky 2,3). Patří k nim hypoplastický mozeček, obličejové dysmorfismy (tj. velké, dysplastické uši), vpáčené bradavky a abnormální rozložení tukové tkáně nad hýžděmi nebo v suprapubické oblasti, které může s věkem ustoupit (14,21,29-32). U pacientů je popisováno společenské a veselé chování. Projevy jsou velmi variabilní, i když strabismus lze pozorovat u více než 70 % postižených pacientů (21,23,33-35). Invertované bradavky a abnormální tukové polštářky jsou pozorovány asi u 25-50 % pacientů (36).

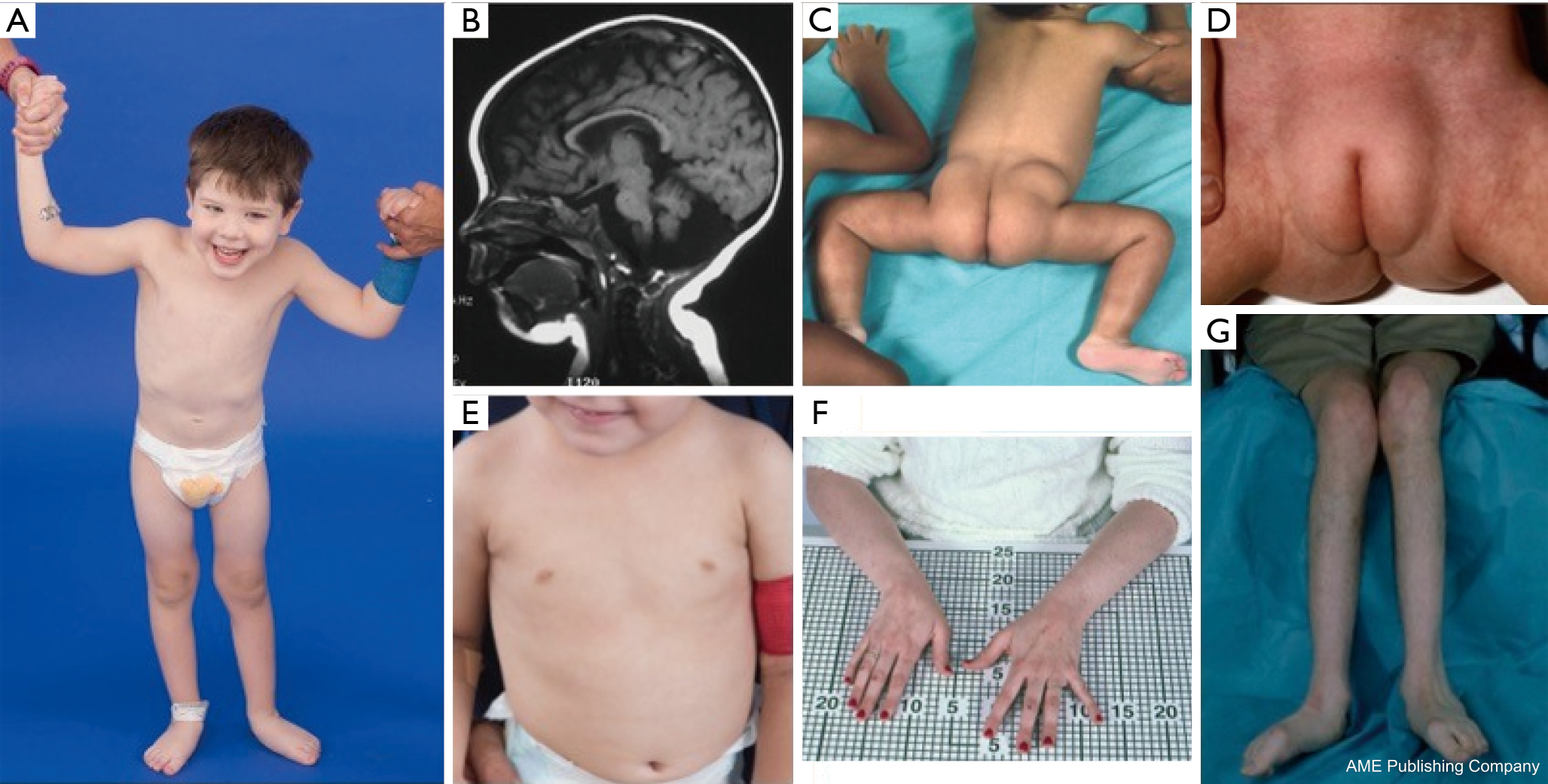
V dětství se u postižených jedinců může vyvinout pigmentová retinitida, příhody podobné mrtvici a záchvaty, opoždění řeči a motoriky a periferní neuropatie. Konstitučně pacienti běžně neprospívají v důsledku poruch krmení a trávicího traktu a mají globální vývojové opoždění. Mohou být pozorovány zvýšené jaterní transaminázy bez klinických důsledků, které se obvykle normalizují do 5 let věku s občasnými výkyvy při onemocnění (21,24). Jaterní biopsie je u CDG indikována zřídka, pokud není podezření na jaterní fibrózu (1). Klinická hypotyreóza je vzácná, ale pacienti s CDG by si měli nechat změřit hormony štítné žlázy a volný T4, což může ukázat nízkou hladinu globulinu vázajícího štítnou žlázu (TBG) a přechodné zvýšení hormonu stimulujícího štítnou žlázu (TSH) (37). Malformace jater a žlučových cest nebyly u pacientů s PMM2-CDG hlášeny.
Dospělí s PMM2-CDG se mohou dožít 7. nebo 8. dekády se stabilním kognitivním opožděním, periferní neuropatií a progresivní hrudní a spinální kyfoskoliózou s osteopenií nebo osteoporózou (34). Stále častěji rozpoznávaným příznakem je mozečková ataxie spolu s multisystémovým postižením (38-40). Endokrinní abnormality zahrnující hyperprolaktinémii, uvolňování růstového hormonu s hyperglykémií, inzulinovou rezistenci a hyperinzulinemickou hypoglykémii (41,42). U postižených žen může hypogonadotropní hypogonadismus vést k absenci sekundárního pohlavního vývoje nebo k absenci vaječníků (41,43,44). Pacienti mohou být vystaveni zvýšenému riziku trombózy v důsledku snížení sérových koagulačních faktorů včetně faktorů IV, IX a XI, antitrombinu III, proteinu C a proteinu S (29).
MPI-CDG (CDG-Ib, deficit mannosafosfát izomerázy)
MPI-CDG je unikátní tím, že postižení pacienti mají malé nebo žádné neurologické postižení a některé projevy onemocnění jsou léčitelné perorální manózou (2). Příznaky jsou převážně jaterně-střevní bez dysmorfických rysů nebo opoždění kognitivních funkcí. U pacientů se typicky vyskytuje opakované zvracení, významná hypoglykémie, neprospívání, potenciálně život ohrožující enteropatie s nedostatkem bílkovin, fibrotické změny jater a dilatace žlučových cest (45-51). Pacienti mají zvýšené riziko trombotických příhod kvůli nízkým sérovým koncentracím proteinů C a S a antitrombinu III.
ALG6-CDG (deficit glukosyltransferázy 1)
ALG6-CDG je druhý nejčastější defekt N-glykosylace charakterizovaný podobným, ale mírnějším fenotypem než PMM2-CDG. Pacienti s ALG6-CDG neprospívají, mají opožděný vývoj, hypotonii, záchvaty, strabismus, ataxii, koagulopatii a obličejový dysmorfismus (tj. nízko posazené uši, hypertelorismus a makroglosii). Podobně jako u MPI-CDG se u nich může vyskytnout také enteropatie se ztrátou bílkovin. Postižení pacienti mohou mít navíc abnormality skeletu včetně brachydaktylie a malformací prstů a skoliózy. Postižení pacienti obvykle nemají pigmentovou retinitidu ani hypoplazii mozečku (52).
O-vázané defekty glykosylace a kombinované defekty N- a O-vázané glykosylace
Vzhledem k významné přítomnosti O-glykanů v proteinech obsahujících mucin včetně glykosaminoglykanů (GAG) a epiteliálních povrchů (53) vedou poruchy syntézy GAG typicky ke kosterním dysplaziím nebo onemocnění pojivové tkáně. U postižených pacientů se kromě neurologických příznaků mohou vyskytovat muskuloskeletální, kožní a kloubní abnormality (např. kloubní laxita, mnohočetné exostózy, chondro/osteosarkomy) (54-56). Například N-acetylgalaktosaminyltransferáza 3 (GALNT3) O-glykosyluje fosfaturický hormon FGF23, čímž zabraňuje proteolytickému štěpení a umožňuje jeho intaktní sekreci. Deficit GALNT3 vede k familiární nádorové kalcinóze, která je charakterizována hyperfosfatémií a ektopickými kalcifikacemi (57,58).
Defekty glykosylace lipidů a biosyntézy GPI kotev
Glykosfingolipidy a jejich sialylované deriváty, gangliosidy, jsou exprimovány především neurony. Defekty v odbourávání gangliosidů vedou k jejich hromadění a dobře charakterizovaným lysozomálním střádacím chorobám. Na opačném konci jsou defekty v biosyntéze gangliosidů, jako jsou ST3GAL5-CDG a B4GALNT1-CDG, které jsou mimořádně vzácné a vedou k závažným neurodegenerativním onemocněním. Pacienti se mohou projevovat spastickou paraplegií, těžkým opožděním intelektu, epilepsií a neurologickými příznaky, včetně dysplazie skeletu, dysmorfních rysů a abnormální pigmentace kůže (59,60).
Mutace v mnoha genech v rámci biosyntetické dráhy GPI kotvy způsobují různé mnohočetné vrozené anomálie, intelektuální postižení a epilepsii. Nejlépe charakterizovaný defekt biosyntézy GPI, X-vázaný deficit PIGA, se projevuje dětskými křečemi s hypsarytmií, hypotonií, četnými mozkovými abnormalitami a obličejovým dysmorfismem. Pacienti mohou mít také různá onemocnění kůže, jater, srdce a ledvin (61-69). Některé mutace v rámci PIGA způsobují fenotypově odlišné onemocnění paroxysmální noční hemoglobinurii (PNH), získanou poruchu selhání kostní dřeně (70,71).
Diagnostika
Při klinickém podezření na CDG je prvním krokem objednání biochemického vyšetření CDG v plazmě nebo séru, včetně vyšetření CDT a N-glykanu. Analýza CDT a N-glykanu v séru může odhalit pouze defekty N-glykosylace, proto by nebyla užitečná pro odlišení izolovaných defektů O-glykosylace nebo GPI kotvy. Analýza izoforem transferinu byla původně získána izoelektrickou fokusací transferinu, protože porucha syntézy N-glykanu způsobuje částečný nedostatek kyseliny sialové, což mění náboj na sérovém transferinu a následně jeho katodovou migraci na elektroforetickém poli. Analýza transferinu a N-glykanu založená na hmotnostní spektrometrii však nyní z velké části nahradila izoelektrickou fokusaci tím, že identifikuje specifické změny oligosacharidů podle hmotnosti a náboje (72).
N-vázané defekty glykosylace proteinů
Výsledky CDT transferinu v séru jsou uváděny jako poměr mono-oligosacharidů/di-oligosacharidů transferinu, a-oligosacharid/di-oligosacharidový transferin, poměr tri-sialo/di-oligosacharidový transferin, poměr apolipoprotein CIII-1/apolipoprotein CIII-2 a poměr apolipoprotein CIII-0/apolipoprotein CIII-2. Tyto kvantitativní výsledky přijdou také s interpretací vzorce nálezů.
Typ I vzorce transferinového CDT je charakterizován zvýšenými di- a asialotransferinovými pásy a ukazuje na defekty v syntéze N-glykanu v cytosolu nebo endoplazmatickém retikulu. Vzor typu II je charakterizován zvýšenými di- a asialotransferrinovými pásy a tri- a/nebo monosialotransferrinovými pásy a ukazuje na defekty ve zpracování N-glykanů v Golgiho aparátu (73).
Pokud je zjištěn vzor CDT transferinu v séru typu I, měl by být v popředí diferenciální diagnostiky deficit PMM2 nebo MPI, protože PMM2-CDG je nejčastější CDG a MPI-CDG je léčitelný a potenciálně smrtelný, pokud se neléčí. K rozlišení diagnóz by mělo být provedeno profilování N-glykanů, molekulární sekvenování nebo enzymatické vyšetření. Diagnóza PMM2-CDG nebo MPI-CDG se potvrdí molekulárním testováním, které prokáže bialelické patogenní varianty v PMM2 nebo MPI, a následným vyšetřením aktivity enzymu PMM nebo MPI v leukocytech nebo fibroblastech, pokud není patogenita genetických variant jistá. Analýza N-glykanů nebo molekulární analýza by odlišila většinu ALG-CDG od PMM2 nebo MPI-CDG (15).
Sérový transferinový vzor CDT typu II ukazuje na defekty Golgiho, jako je nedostatek N-acetylglukosaminltransferázy (GnT) II (CDG typu IIA, MGAT2-CDG). Analýza izoforem apolipoproteinu CIII (Apo-CIII) je doplňkovým testem pro profil CDT typu II, protože měří defekty glykosylace mucinu typu O v Golgiho aparátu. Citlivost CDT nebo Apo-CIII při detekci CDG typu II je omezená. Proto by mělo být provedeno profilování N-glykanů a O-glykanů a molekulární panel nebo sekvenování exomu, pokud jsou tyto klinické testy k dispozici. Glykosylační vzorce transferinu se mohou sporadicky normalizovat, proto může být u pacientů s vysokým indexem podezření indikováno opakované testování. Falešně pozitivní výsledky mohou být získány u pacientů s akutní krizí dědičné intolerance fruktózy, galaktosemií, akutním onemocněním jater a některými bakteriálními infekcemi. Žádný z biochemických testů CDG nedokáže vyšetřit všechny CDG, a proto i v případě normálních výsledků screeningu může být při silném klinickém podezření provedeno vyšetření molekulárního genového panelu nebo sekvenování exomu. Naopak biochemické a funkční potvrzení molekulárně genetických nálezů je rovněž nezbytné, protože většina pacientů s CDG nese alespoň jednu mírnou a často novou missense mutaci.
O-vázané glykosylační defekty a kombinované N- a O-vázané glykosylační defekty
Diagnostika závisí na molekulárním sekvenování, protože analýza izoforem transferinu by neodhalila izolované O-glykosylační defekty. Kombinované defekty N- a O-glykosylace lze zjistit pomocí CDT, analýzy ApoCIII a analýzy N-glykanu a O-glykanu v plazmě.
Defekty glykosylace lipidů a biosyntézy GPI kotev
Průtoková cytometrie krevních granulocytů měří expresi proteinů kotvených GPI, jako jsou CD16 a CD24, na povrchu buněk. Analýza průtokové cytometrie bílých krvinek nebo červených krvinek na přítomnost určitých povrchových proteinů kotvených GPI je klinicky dostupná jako test na PNH v důsledku získaných mutací v genu PIGA. Test PNH může odhalit abnormality u jiných deficitů kotvy GPI, ale diagnostika většinou závisí na molekulární analýze.
Molekulární analýza
Nejvyšší diagnostickou výtěžnost pro CDG představuje panel genového sekvenování založené na sekvenování nové generace nebo klinické sekvenování exomu (CES). Genetické sekvenování prověřuje nukleotidovou sekvenci neboli „písmenný“ pravopis genů, aby se zjistilo, zda existuje změna, která ovlivňuje funkci genu. Lidský genom se skládá ze 3 milionů nukleotidů, ale pouze 1-2 % z nich, tzv. exony, jsou přeloženy do funkčního proteinového produktu. Zbývající nekódující DNA proložená mezi exony, která se nepřekládá, se nazývá introny (74). CES zkoumá téměř všechny známé exony z přibližně 20 000 genů v lidském genomu, které tvoří menšinu genetického materiálu v chromozomech, ale s největší pravděpodobností obsahují varianty způsobující onemocnění (patogenní). CES může zahrnovat také sekvenování mitochondriální DNA (mtDNA), které zkoumá malou, extranukleární, kruhovou DNA umístěnou v mitochondriích, která se dědí výhradně po matce.
Možné výsledky CES zahrnují pozitivní, negativní a varianty neznámého významu. Pozitivní výsledek znamená, že jsou identifikovány známé varianty způsobující onemocnění (tj. patogenní), načež lze diskutovat o diagnóze, přirozeném průběhu, prognóze, riziku recidivy a možnostech léčby. Negativní výsledek znamená, že nebyly identifikovány žádné zjistitelné patogenní varianty. Varianty neznámého významu (tzv. VUS) znamenají, že sice byly identifikovány genetické změny, ale o konkrétní genetické změně není dostatek informací, aby bylo možné definitivně určit, zda je příčinou onemocnění. Odchylky v DNA každého jedince se očekávají, proto může laboratorní a klinická interpretace výsledků pomoci, pokud budou současně testovány vzorky rodičů za účelem porovnání. Diagnostická výtěžnost CES se odhaduje na 30-35 % a v průběhu času se zvyšuje s tím, jak postupuje objevování genů a znalosti o lidském genomu (75-77). CES je stále častěji objednáván jako široký genetický test první volby vzhledem k rychlé době provedení a nízkým relativním nákladům vzhledem k množství analyzovaných genetických informací. Mezi omezení CES patří nedostatečná 100% citlivost, neschopnost odhalit určité typy genetických změn (např. delece, duplikace, trinukleotidové repetice, hluboké intronické mutace nebo metylační defekty) a skutečnost, že diagnóza nemusí poskytnout další informace o onemocnění nebo změnit management.
Při hlášení CES mohou být náhodně zjištěné patogenní varianty v genech spojených s dobře známými genetickými stavy hlášeny jako sekundární nálezy (78). Tento seznam doporučených onemocnění je sestaven kurátorem American College of Medical Genetics (ACMG). Zákon o nediskriminaci genetických informací (Genetic Information Nondiscrimination Act, GINA) je důležitým faktorem při rozhodování, zda se rozhodnout pro učení náhodných nálezů nebo ne (79). GINA chrání jednotlivce před zneužitím genetických informací v oblasti zdravotního pojištění a zaměstnání, nikoli však životního pojištění. GINA chrání následující genetické informace: rodinnou anamnézu, testování nosičství, prenatální genetické testování, testování náchylnosti a prediktivní testování a analýzu nádorů nebo jiné hodnocení genů, mutací nebo chromozomálních změn.
Management
Management CDG závisí do značné míry na konkrétních příznacích jedince. Mezi opakující se příznaky u pacientů s CDG patří neprospívání, globální opoždění vývoje, zvracení, příhody podobné mozkové mrtvici a abnormality skeletu. Často se také vyskytuje klinická nebo subklinická koagulopatie, endokrinopatie, hepatopatie a srdeční vady. Doporučují se základní laboratorní testy ke zjištění rozsahu onemocnění a rutinní sledování, zejména u PMM2-CDG. Patří k nim jaterní funkční testy, sérový albumin, funkční testy štítné žlázy včetně volného T4, protein C, protein S, antitrombin III, faktor IX, vyšetření moči a gonadotropiny a růstový hormon v séru.
Doporučené zobrazovací vyšetření zahrnuje echokardiogram, ultrazvuk ledvin, kostní věk, oftalmologické vyšetření k posouzení čočky, sítnice, pohyblivosti oka a nitroočního tlaku. Pokud není uvedeno jinak, doporučuje se u dospělých a dětí postižených CDG rutinní očkování. Po očkování by měly být získány titry protilátek, protože pacienti mohou mít suboptimální imunogenní odpověď. Profylaktické doplnění koagulačních faktorů před jakýmkoli chirurgickým zákrokem může být nezbytné, pokud na počátku existuje jejich nedostatek.
Mělo by být provedeno klinicko-genetické vyšetření, aby se prodiskutovaly dědičné aspekty CDG, a také vytvoření lékařského domova pro tyto složité pacienty. Lékařským domovem je obvykle oddělení biochemické genetiky, ačkoli v případě, že specializované oddělení biochemické genetiky není k dispozici, plní tuto funkci také oddělení genetiky, neurovývoje nebo neurologie. Často je nutné odeslání ke specialistovi na gastroenterologii, hematologii, endokrinologii, nutriční podporu, logopedii, ergoterapii, fyzikální terapii a terapii výživy, ortopedii a rehabilitační medicínu.
Cílené terapie a prognóza
Léčba většiny typů CDG je až na výjimky převážně podpůrná. MPI-CDG je ze všech typů CDG nejúčinněji léčitelná. Perorální manóza je přeměněna na manóza-6-fosfát intracelulárními hexokinázami, čímž se obejde enzymatický blok a vznikne deficitní substrát. Suplementace manózy obvykle začíná dávkou 1 g/kg tělesné hmotnosti denně, rozdělenou do 4-6 dávek denně. Zatímco potenciálně život ohrožující enteropatie s nedostatkem bílkovin reaguje na léčbu manózou obzvláště dobře, onemocnění jater u MPI-CDG může dále progredovat. Klinické příznaky se rychle zlepšují a CDT transferinu se během několika měsíců normalizuje, ačkoli jaterní onemocnění může při léčbě dále progredovat (45,80,81).
Při podávání manózy během těhotenství je třeba postupovat opatrně, protože podávání manózy u březích hypomorfních myších modelů s fosfomanózovou izomerázou vedlo k embryonální letalitě a slepotě jejich mláďat (82). Kromě toho byla intravenózní manóza spojena se sníženým vědomím a záchvaty, které ustoupily při podání glukózy (83).
Léčba PMM2-CDG je převážně podpůrná a vychází ze symptomatologie. V současné době se však připravují klinické studie týkající se substituční léčby manóza-1-fosfátem.
U ostatních CDG byly zkoušeny různé perorální jednoduché cukry s cílem teoreticky zlepšit hypoglykosylaci. U SLC35C1-CDG byla zkoušena fukóza a u PGM1-CDG a SLC35A2-CDG galaktóza se smíšenými výsledky (84). Bylo prokázáno, že D-galaktóza v dávce 1,0-2,5 g/kg/den (max. 50 g) zlepšuje hypoglykémii, koagulopatii a endokrinopatii u PGM1-CDG (85,86). Bylo rovněž prokázáno, že galaktóza zlepšuje endokrinopatii a koagulopatii u TMEM165-CDG (87) a SLC39A8-CDG. Výrazné klinické zlepšení bylo zaznamenáno také u pacientů se SLC39A8-CDG, kteří dostávali 15-20 mg/kg/den MnSO4 (88). Probíhají klinické studie zaměřené na užitečnost N-acetylmannosaminu (ManNAc) u GNE-CDG (89) a několik předklinických studií probíhá i u jiných CDG (90).
I přes pokroky v medicíně existuje u dětí s CDG během prvního roku života významná úmrtnost na multiorgánové selhání nebo závažné infekce (91). U kojenců s CDG se může projevit fulminantní multiorgánové onemocnění, neřešitelné záchvaty nebo těžká hypoalbuminemie progredující do anasarky. Někteří pacienti reagují na agresivní diurézu a náhradu albuminu, zatímco jiní jsou vůči léčbě refrakterní. Bylo prokázáno, že butyrát sodný zlepšuje kontrolu záchvatů u CAD-CDG a PIGM-CDG (92). Bylo také prokázáno, že ketogenní dieta snižuje frekvenci záchvatů v některých případech PIGA-CDG (93). Během epizod podobných cévní mozkové příhodě může pomoci intravenózní hydratace a udržování normální hladiny glukózy v krvi, zatímco je vyloučena základní cévní trombotická nebo krvácivá etiologie.
S nástupem technik editace genomu a lepším pochopením mechanismu onemocnění zahrnutých do diagnostického deštníku CDG zůstává budoucnost vývoje cílené terapie slibná.
Poděkování
Rádi bychom poděkovali Lynne Wolfe, ARNP a Donně Krasnewich, MD, PhD za poskytnutí klinických fotografií získaných v rámci projektu Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). Rádi bychom také poděkovali Jenny Thiesové, MS, LGC, za její odborné znalosti v oblasti genetického poradenství.
Financování: IJ Chang je podporován National Institutes of Health T32GM007454.
Poznámka pod čarou
Konflikty zájmů:
Informovaný souhlas: Od pacientů byl získán písemný informovaný souhlas se zveřejněním tohoto rukopisu a všech doprovodných obrázků.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutace v PMM2, které způsobují vrozené poruchy glykosylace typu Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG nomenclature: time for a change! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways (Řešení poruch glykosylace: základní přístupy odhalují komplikované cesty). Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Co je nového v CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Congenital disorders of glycosylation. Část I. Defekty N-glykosylace proteinů. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracelulární funkce N-vázaných glykanů. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Klinické, laboratorní a molekulární nálezy a údaje z dlouhodobého sledování u 96 francouzských pacientů s PMM2-CDG (fosfomannomutáza 2 – vrozená porucha glykosylace) a přehled literatury. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG in South Africa: Genotype-Phenotype Description of Five Novel Patients: Genotype-Phenotype Description of Five Novel Patients. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Different glycosyltransferases are differentially processed for secretion, dimerization, and autoglycosylation. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In: Glykosyntéza v organismu: Mora-Montes H. Editor. Glycans: Vrozené poruchy glykosylace: Biochemie, charakterizace a aplikace. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Kožní projevy u CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF systém u dětí s vrozenými poruchami glykosylace. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertální růst u vrozené poruchy glykosylace typu Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Klinické a biochemické charakteristiky vrozené poruchy glykosylace typu Ic, prvního rozpoznaného defektu endoplazmatického retikula v syntéze N-glykanu. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Vrozené poruchy glykosylace: přehled jejich molekulárních základů, klinických projevů a specifické terapie. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1– nedávno objevené dědičné metabolické onemocnění. Mnohočetné orgánové projevy, incidence 1/80 000, obtížně léčitelné. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Těžká přechodná ischemie myokardu způsobená hypertrofickou kardiomyopatií u pacienta s vrozenou poruchou glykosylace typu Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Konotunkční srdeční vady u tří pacientů s vrozenou poruchou glykosylace typu Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Perikardiální a abdominální akumulace tekutiny u vrozené poruchy glykosylace typu Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Klinické rysy u dospělých s vrozenou poruchou glykosylace typu Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Syndrom glykoproteinů s deficitem sacharidů. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Klinické a molekulární rysy vrozené poruchy glykosylace u pacientů se sialotransferinovým vzorem typu 1 a různým etnickým původem. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modifikace plášťového proteinu potyviru Plum pox virus zvyšuje virovou infekci. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Scandinavian CDG-Ia patients: genotype/phenotype correlation and geographic origin of founder mutations. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 francouzských dospělých pacientů s vrozenou poruchou glykosylace PMM2: výsledek klasického dětského fenotypu a zobrazení fenotypu s pozdním nástupem. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinální charakteristiky vrozené poruchy glykosylace PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Thyroid function in PMM2-CDG: diagnostic approach and proposed management. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Nové poruchy v metabolismu sacharidů: vrozené poruchy glykosylace a jejich dopad na endokrinní systém. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endokrinologie syndromu glykoproteinového deficitu sacharidů typu 1 od narození do dospívání. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadální funkce a glykoproteinové hormony u syndromu carbohydrate-deficient glycoprotein (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Klinické spektrum deficitu fosfomanózové izomerázy s hodnocením léčby manózou u CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Deficit fosfomanózové izomerázy a léčba manózou. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinzulinemická hypoglykémie jako prezentační příznak při deficitu fosfomanózní izomerázy: Nový projev syndromu glykoproteinového deficitu sacharidů léčitelný manózou. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG6 defektní vrozená porucha glykosylace. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. hlen dýchacích cest: Od produkce k sekreci. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparan sulfát ve vývoji, růstu a patologii skeletu: případ dědičné mnohočetné exostózy. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Různé nezánětlivé muskuloskeletální stavy. Hyperfosfatemická familiární tumorózní kalcinóza (FGF23, GALNT3 a αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Ablace genu Galnt3 vede k nízkým koncentracím intaktního fibroblastového růstového faktoru 23 (Fgf23) a hyperfosfatemii navzdory zvýšené expresi Fgf23. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Mutace v enzymu pro biosyntézu gangliosidů, ST3GAL5, vede k syndromu solného & pepře, neurokutánní poruše se změněnou glykolipidovou a glykoproteinovou glykosylací. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. Mutace PIGA způsobují epileptické encefalopatie s časným nástupem a charakteristickými rysy. Neurologie. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorfní promotorová mutace v PIGM způsobuje dědičný deficit glykosylfosfatidylinositolu. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorfní mutace v PGAP2, kódujícím GPI-anchor-remodeling protein, způsobují autozomálně recesivní mentální postižení. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutace v genu PIGO, členu dráhy syntézy GPI kotev, způsobují hyperfosfatázii s mentální retardací. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutace v genu pro glykosylfosfatidylinositol PIGL způsobují syndrom CHIME. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J 1994;13:110-7.
- Brodsky RA. Paroxysmální noční hemoglobinurie. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. Vliv hmotnostní spektrometrie na diagnostiku vrozených poruch glykosylace. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomálně recesivní dilatační kardiomyopatie způsobená mutací DOLK je důsledkem abnormální O-mannosylace dystroglykanu. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distribuce exonů a intronů v lidském genomu. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Proudy v současné etice. GINA, ADA a genetická diskriminace v zaměstnání. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Recurrent thrombo-embolism in a child with a congenital disorder of glycosylation (CDG) type Ib and treatment with mannose. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Rozvoj jaterního onemocnění navzdory léčbě manózou u dvou pacientů s CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Záchvaty a stupor během intravenózní léčby manózou u pacienta s CDG syndromem typu 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Suplementace fukózou u deficitu adheze leukocytů typu II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Dědičný deficit glykosylfosfatidyl inositolu: léčitelná CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Perorální suplementace D-galaktózy u PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Suplementace galaktózy u pacientů s TMEM165-CDG zachraňuje glykosylační defekty. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8 deficience: biochemická korekce a významné klinické zlepšení pomocí terapie manganem. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Human glycosylation disorders. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogenní dieta – nová léčba časné epileptické encefalopatie způsobené nedostatkem PIGA. Brain Dev 2016;38:848-51.