- Resumen
- Epidemiología
- Clasificación bioquímica y nomenclatura
- Genética
- Patofisiología
- Los defectos de glicosilación de proteínas ligadas al N
- Los defectos de glicosilación ligados a O y los defectos combinados de glicosilación ligados a N y O
- Defectos de glicosilación de lípidos y de biosíntesis de anclajes GPI
- Manifestaciones clínicas
- Defectos de la glicosilación de proteínas ligadas al N
- PMM2-CDG (CDG-Ia, deficiencia de PMM2)
- MPI-CDG (CDG-Ib, deficiencia de manosa-fosfato isomerasa)
- ALG6-CDG (deficiencia de glucosiltransferasa 1)
- Defectos de glicosilación ligados a O y defectos combinados de glicosilación ligados a N y O
- Defectos de glicosilación de lípidos y de biosíntesis de anclajes GPI
- Diagnóstico
- Defectos de glicosilación de proteínas ligadas a N
- Defectos de glicosilación ligados a O y defectos de glicosilación combinados ligados a N y O
- Defectos de glicosilación de lípidos y de biosíntesis de anclajes GPI
- Análisis molecular
- Manejo
- Terapias dirigidas y pronóstico
- Agradecimientos
- Nota
Resumen
La glicosilación es el proceso de adición de residuos de azúcar a las proteínas y a los lípidos en diferentes vías celulares. Los trastornos congénitos de la glicosilación (CDG) son un grupo genética y clínicamente heterogéneo de más de un centenar de enfermedades causadas por defectos en varios pasos a lo largo de las vías de síntesis o modificación de glicanos. La mayoría de estas enfermedades monogénicas son de herencia autosómica recesiva, pero también se han descrito formas autosómicas dominantes y ligadas al cromosoma X.
Las CDG se presentan típicamente con manifestaciones multisistémicas, más comúnmente retraso en el desarrollo, retraso en el crecimiento, hipotonía, anomalías neurológicas, hepatopatía y coagulopatía. Los individuos afectados también pueden presentar enfermedades oculares, cutáneas y cardíacas, así como dismorfismos faciales. Aunque se observan cambios neurológicos y retrasos cognitivos en la mayoría de los individuos afectados, hay ciertos casos e incluso tipos que no tienen manifestaciones neurológicas. Dada la amplia etiología clínica y genética de la CDG, el diagnóstico clínico se basa en un alto índice de sospecha en la enfermedad multisistémica.
El análisis de la transferrina deficiente en carbohidratos en suero (CDT) es la prueba de cribado de primera línea en pacientes con sospecha de CDG, pero su detección está limitada a los defectos de N-glicosilación con deficiencias de ácido siálico. Las pruebas de siguiente línea incluyen el análisis de glicanos ligados a dolicol y las pruebas genéticas. El diagnóstico temprano de este grupo de enfermedades de crecimiento exponencial es importante, ya que algunas CDG son tratables. El tratamiento de los defectos de glicosilación es principalmente de apoyo, aunque existen terapias dirigidas para MPI-CDG, SLC35C1-CDG, PIGM-CDG y PGM1-CDG. Los detalles sobre estos tratamientos se encuentran en la sección «Terapias dirigidas y pronóstico» más adelante. Esta revisión se centrará en los tipos más comunes de CDG con fenotipos o tratamientos reconocibles, siendo el público objetivo los proveedores de atención primaria.
Epidemiología
La incidencia y la prevalencia de todos los tipos de CDG en conjunto no se han establecido bien, aunque se han notificado pacientes en todo el mundo de casi todos los orígenes étnicos y ambos sexos están igualmente afectados. La prevalencia estimada en las poblaciones europeas y afroamericanas es de 1/10.000, basándose en las frecuencias de portadores de variantes patogénicas conocidas en 53 genes (1-4). La prevalencia del CDG más comúnmente diagnosticado, el PMM2-CDG, oscila entre 1/20.000 en poblaciones holandesas y 1/77.000 en Estonia, basándose en informes aislados (5,6). Hasta la fecha, se han notificado menos de 100 casos para la mayoría de los tipos de CDG.
Clasificación bioquímica y nomenclatura
En términos generales, los CDG se clasifican actualmente en cuatro categorías-(I) glicosilación ligada a N, (II) glicosilación ligada a O, (III) glicosilación combinada de N y O/múltiple, y (IV) defectos de biosíntesis de anclaje de lípidos y glicosilfosfatidilinositol (GPI).
El defecto de glicosilación de proteínas ligadas al N PMM2-CDG, anteriormente conocido como CDG tipo Ia, fue el primer CDG reportado por Jaeken en 1980 y sigue siendo con mucho el CDG más común hasta la fecha (7). El PMM2-CDG se denominó inicialmente «síndrome de glicoproteínas deficientes en carbohidratos» debido a las múltiples anomalías de las glicoproteínas séricas observadas mediante el enfoque isoeléctrico de la transferrina sérica en los individuos afectados. Históricamente, los CDG se clasificaban según los patrones de análisis de las isoformas de la transferrina: los patrones de tipo I se atribuían a defectos de ensamblaje y transferencia de los glicanos ligados al dolicol que se localizaban en el citoplasma o en el RE, y los patrones de tipo II se atribuían a defectos de procesamiento en el aparato de Golgi. A partir de este punto de ramificación, los CDGs se nombraron alfabéticamente en orden de descubrimiento.
Con la llegada de los diagnósticos moleculares generalizados, la nomenclatura de los CDGs se actualizó en 2008 para especificar la etiología molecular de la enfermedad, reflejando el crecimiento exponencial de las vías y trastornos que no encajaban claramente en las categorías dicotómicas previamente establecidas. En la actualidad, la nomenclatura de los CDG se denota por el nombre del gen afectado (en mayúsculas, nombres de genes en www.genenames.org), seguido de -CDG (por ejemplo, PMM2-CDG) (8).
Genética
La gran mayoría de los trastornos congénitos de la glicosilación se heredan de forma autosómica recesiva, con una mutación heredada de cada progenitor asintomático (portador). Es necesario realizar pruebas moleculares, normalmente con métodos de secuenciación de nueva generación, para establecer un diagnóstico genético. Las pruebas de los padres para la variante conocida pueden confirmar la herencia frente a la aparición de novo. En el caso de la herencia autosómica recesiva, el riesgo de recurrencia en los hermanos y en cada embarazo de un individuo afectado es del 25% en caso de estar afectado, del 50% en caso de ser portador asintomático y del 25% en caso de no estar afectado.
Un puñado de CDG tienen una herencia autosómica dominante (ligada al N: GANAB-CDG, PRKCSH-CDG; ligada a la O: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Menos son ligados al X (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). La mayoría de las formas dominantes y algunas ligadas al X de CDG se deben a mutaciones de novo. Las enfermedades y los genes específicos se describen a continuación en la sección «fisiopatología».
Los datos de las mutaciones de todos los genes publicados para CDG están disponibles en la base de datos de mutaciones genéticas humanas (http://www.hgmd.cf.ac.uk/ac/index.php). La información sobre variantes genéticas específicas está disponible en la Leiden Open Variation Database con herramientas integradas de patogenicidad in silico (http://www.lovd.nl/3.0/home). Se pueden encontrar sinopsis clínicas para genes específicos en la Online Mendelian Inheritance in Man (http://www.omim.org/) o en un ámbito más limitado en GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Dado el pequeño número de pacientes afectados para la mayoría de los subtipos de CDG, la correlación genotipo-fenotipo es difícil de establecer.
Patofisiología
Hasta la fecha se han descrito más de 130 tipos de CDG (9,10). Dada la presencia ubicua de vías de glicosilación, los CDG son extremadamente diversos en su patogénesis bioquímica. Numerosas proteínas y lípidos (es decir, esfingolípidos y glicolípidos) sufren glicosilación con monosacáridos y/u oligosacáridos, denominados colectivamente glicanos, en diferentes compartimentos celulares. Sus localizaciones subcelulares son diversas, pero la mayoría de los defectos se producen dentro del RE o del aparato de Golgi. Las características clínicas y la etiología genética de los CDG más comunes por vía se resumen en la Tabla S1.
Entre las proteínas, los glicanos se describen por su unión a la cadena polipeptídica-los N-glicanos se unen al grupo amida de la asparagina (Asn) mientras que los O-glicanos se unen al grupo hidroxilo de la serina o la treonina. La síntesis de los N-glicanos requiere la construcción paso a paso de azúcares ligados a nucleótidos en el citosol, el ensamblaje en el retículo endoplásmico y el procesamiento en el aparato de Golgi. En cambio, la síntesis de O-glicanos requiere el ensamblaje pero no el procesamiento, por lo que los defectos de O-glicosilación se producen predominantemente en el aparato de Golgi.
Los defectos de glicosilación de proteínas ligadas al N
La glicosilación del N implica la unión covalente de estructuras de carbohidratos al grupo amida de la cadena lateral de los residuos de Asn dentro de un sitio aceptor consensuado Asn-X-Ser/Thr, la translocación del polipéptido sustrato al retículo endoplásmico para su remodelación y la posterior modificación de la cadena de N-glicanos dentro del Golgi (11,12). Los defectos en cualquier punto de la vía de síntesis, ensamblaje y procesamiento pueden conducir a la enfermedad clínica.
La PMM2-CDG está causada por variantes patogénicas en el gen de la fosfomanomutasa 2 (PMM2), que conducen a la deficiencia de la enzima PMM2 que cataliza la conversión citosólica de manosa-6-fosfato a manosa-1-fosfato en el segundo paso de la síntesis de la guanosina difosfato (GDP) manosa. La mayoría de los pacientes albergan mutaciones patogénicas de sentido erróneo heterocigotas compuestas (www.lovd.nl/PMM2). La variante patogénica recurrente más común p.Arg141His se encuentra en aproximadamente el 40% de los individuos afectados de ascendencia europea, y p.Phe119Leu también se encuentra con frecuencia en el norte de Europa (1). Se han descrito correlaciones genotipo-fenotipo para la PMM2-CDG (3,13,14).
La PMI-CDG es un trastorno autosómico recesivo causado por variantes patogénicas en el gen de la manosa fosfato isomerasa (MPI) que conducen a una fosfomanosa isomerasa (MPI) deficiente. La MPI normalmente cataliza el primer paso de la síntesis de GDP-manosa (es decir, la conversión de fructosa-6-fosfato en manosa-6-fosfato), pero la fructosa-6-fosfato no se acumula intracelularmente ya que también puede ser metabolizada por la vía glucolítica. Por lo tanto, aunque es bioquímicamente similar a la PMM2-CDG, la MPI-CDG no causa una afectación neurológica y multisistémica tan importante. La CDT es también la prueba de cribado de elección para la MPI-CDG, que muestra un patrón de tipo 1. El diagnóstico puede confirmarse molecularmente o mediante la actividad MPI de fibroblastos/leucocitos.
La ALG6-CDG es una enfermedad recesiva causada por mutaciones en la ALG6, que conducen a la unión anormal de tres moléculas de glucosa a los intermediarios de manosa ligados al dolichol y a la hipoglucosilación posterior de las glicoproteínas séricas (15).
Los defectos de glicosilación ligados a O y los defectos combinados de glicosilación ligados a N y O
La glicosilación O comprende la adición gradual de cadenas de carbohidratos a los residuos de serina, treonina e hidroxilisina de las proteínas por parte de las glicosiltransferasas del aparato de Golgi (16). Se han asociado varios tipos de glicanos ligados a O con enfermedades humanas, denominados por el primer azúcar unido al residuo de aminoácido (17).
Defectos de glicosilación de lípidos y de biosíntesis de anclajes GPI
Los anclajes GPI son glicolípidos que sufren un ensamblaje secuencial en el retículo endoplásmico y modificaciones dentro del Golgi. Los trastornos de la biosíntesis de los anclajes GPI debidos a deficiencias enzimáticas se nombran alfabéticamente por orden de descubrimiento y no cronológicamente por paso de ensamblaje. Una vez sintetizados, los anclajes GPI residen en las membranas plasmáticas y se unen a cientos de proteínas de la superficie celular, realizando una plétora de funciones celulares. La mayoría de estas enfermedades son autosómicas recesivas, con la notable excepción de la deficiencia de PIGA ligada al cromosoma X.
Manifestaciones clínicas
Dada la presencia ubicua de las vías de glicosilación, prácticamente cualquier sistema orgánico puede estar implicado en la CDG, aunque la mayoría de los casos implican anomalías neurológicas. Algunos CDG se presentan con ictiosis, incluyendo MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (Figura 1) y PIGL-CDG (18,19). Casi todos los CDG presentan una enfermedad multisistémica en los primeros años de vida, excepto algunos que sólo afectan a un único sistema orgánico (p. ej, retina en DHDDS-CDG; unión neuromuscular en ALG2-CDG, ALG14-CDG, CFPT1-CDG; cerebro en ST3GAL3-CDG, TUSC3-CDG; piel o músculo esquelético en POGLUT1-CDG, POFUT1-CDG; cartílago en EXT1/EXT2-CDG; hígado en TMEM199-CDG; glóbulos rojos en SEC23B-CDG). La edad de aparición y la gravedad pueden variar desde la letalidad neonatal hasta la edad adulta casi asintomática, y cualquier permutación intermedia. La constelación de síntomas más comúnmente descrita incluye retraso en el desarrollo, retraso en el crecimiento, hipotonía, anomalías neurológicas, hipoglucemia y anomalías hepáticas, oculares, cutáneas, gastrointestinales, inmunológicas, esqueléticas y de coagulación variables (19).
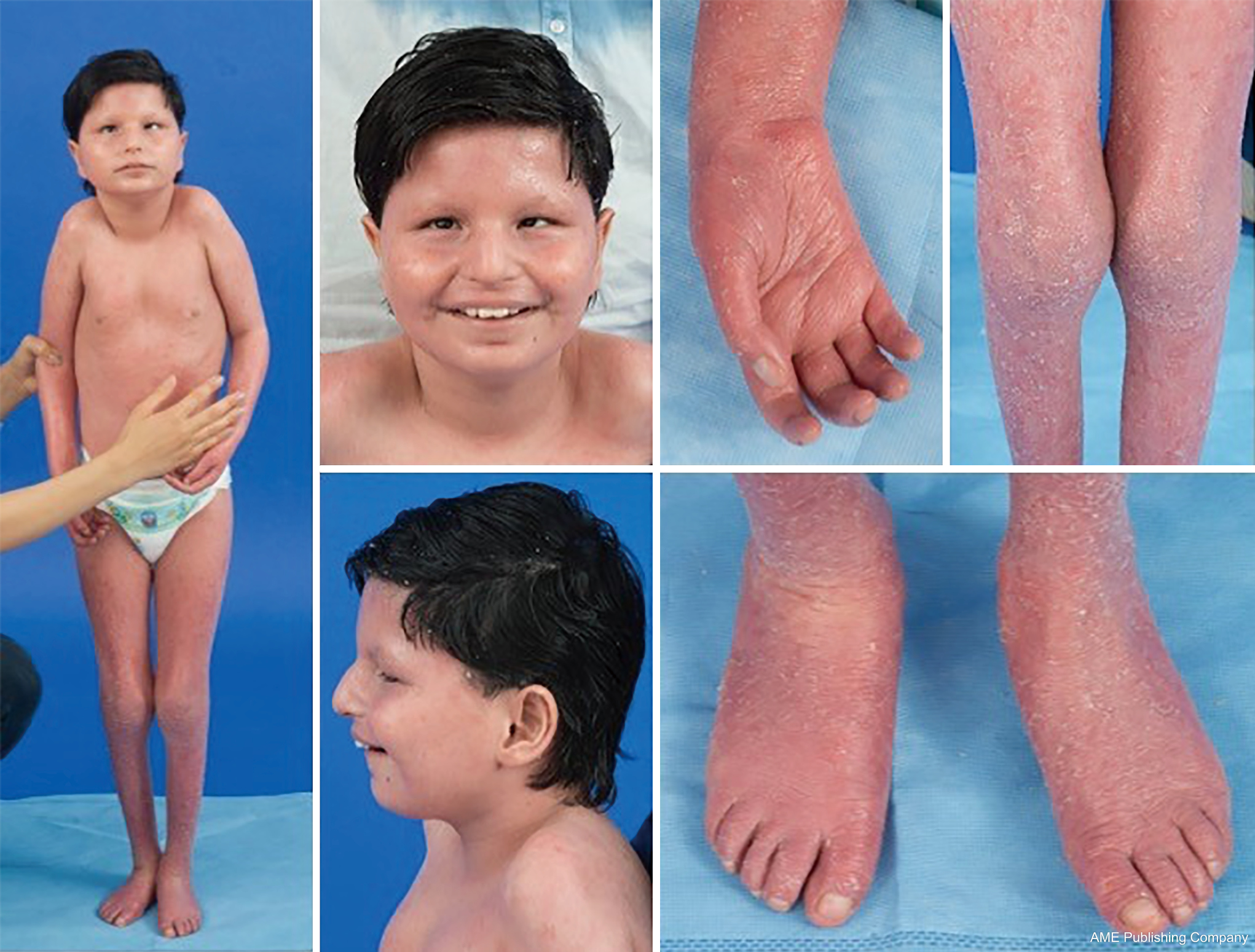
El fenotipo completo de muchos subtipos de CDG aún no se ha delineado completamente debido a la rareza de los casos reportados. Por lo tanto, el CDG debe considerarse en cualquier contexto de enfermedad multisistémica, especialmente en los casos con un componente neurológico o un retraso del desarrollo no específico con una etiología poco clara.
Aunque la fisiopatología de los síntomas de la multitud sigue sin dilucidarse, se ha aclarado la relación entre ciertas vías de glicosilación y síntomas clínicos específicos. Por ejemplo, el retraso en el crecimiento que se observa en muchos tipos de CDG es atribuible a la hipoglicosilación y a la formación alterada de varias glicoproteínas dentro de la vía de crecimiento de la insulina, incluyendo IGF-1, ALS e IGFBP-3 (20). A medida que comprendemos mejor este grupo de trastornos complejos, los CDG se reconocen cada vez más en individuos con diagnósticos elusivos. Los sistemas de órganos implicados en los diferentes CDG se resumen en la Tabla S1. A continuación discutiremos las características clínicas de las formas más comunes y las formas con tratamientos específicos de CDG.
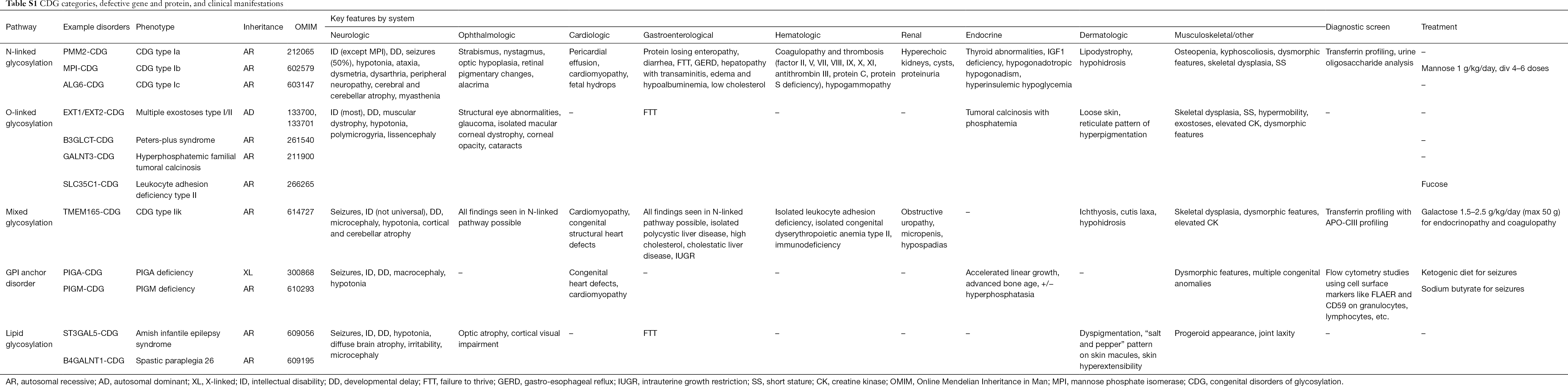
Tabla completa
Defectos de la glicosilación de proteínas ligadas al N
Como el CDG más comúnmente diagnosticado, el fenotipo de los trastornos de la glicosilación ligada al N se anuncia a menudo como la presentación clásica. Sin embargo, el espectro fenotípico de los CDG es bastante diverso, y muchos CDG pueden no presentar los síntomas estereotipados asociados a la PMM2-CDG.
PMM2-CDG (CDG-Ia, deficiencia de PMM2)
PMM2-CDG es el CDG más común, con más de 700 casos registrados en todo el mundo. Se caracteriza por una enfermedad multisistémica grave en la infancia, enfermedad neurológica y retraso del desarrollo en la infancia, y/o discapacidad intelectual estable en la edad adulta (21,22).
En la infancia, la PMM2-CDG se presenta con anomalías neurológicas típicas poco después del nacimiento, a saber, estrabismo y movimientos oculares anormales, hipoplasia cerebelosa, hipotonía, retraso psicomotor, ataxia, hipotonía e hiporreflexia. Los bebés también pueden presentar enfermedad hepática, síndrome nefrótico y quistes renales, derrame pericárdico y miocardiopatía hipertrófica, retraso en el desarrollo y fallo multiorgánico que provoca la muerte durante el primer año de vida en hasta el 20% de los individuos afectados (21,23-28).
Se ha descrito una constelación de rasgos dismórficos en pacientes con PMM2-CDG (Figuras 2,3). Estos incluyen un cerebelo hipoplásico, dismorfismos faciales (es decir, orejas grandes y displásicas), pezones invertidos y una distribución anormal del tejido adiposo sobre las nalgas o la región suprapúbica que puede resolverse con la edad (14,21,29-32). Se ha descrito que los pacientes tienen un comportamiento extrovertido y alegre. La presentación es muy variable, aunque puede observarse estrabismo en más del 70% de los pacientes afectados (21,23,33-35). Se observan pezones invertidos y almohadillas de grasa anormales en aproximadamente el 25-50% de los pacientes (36).

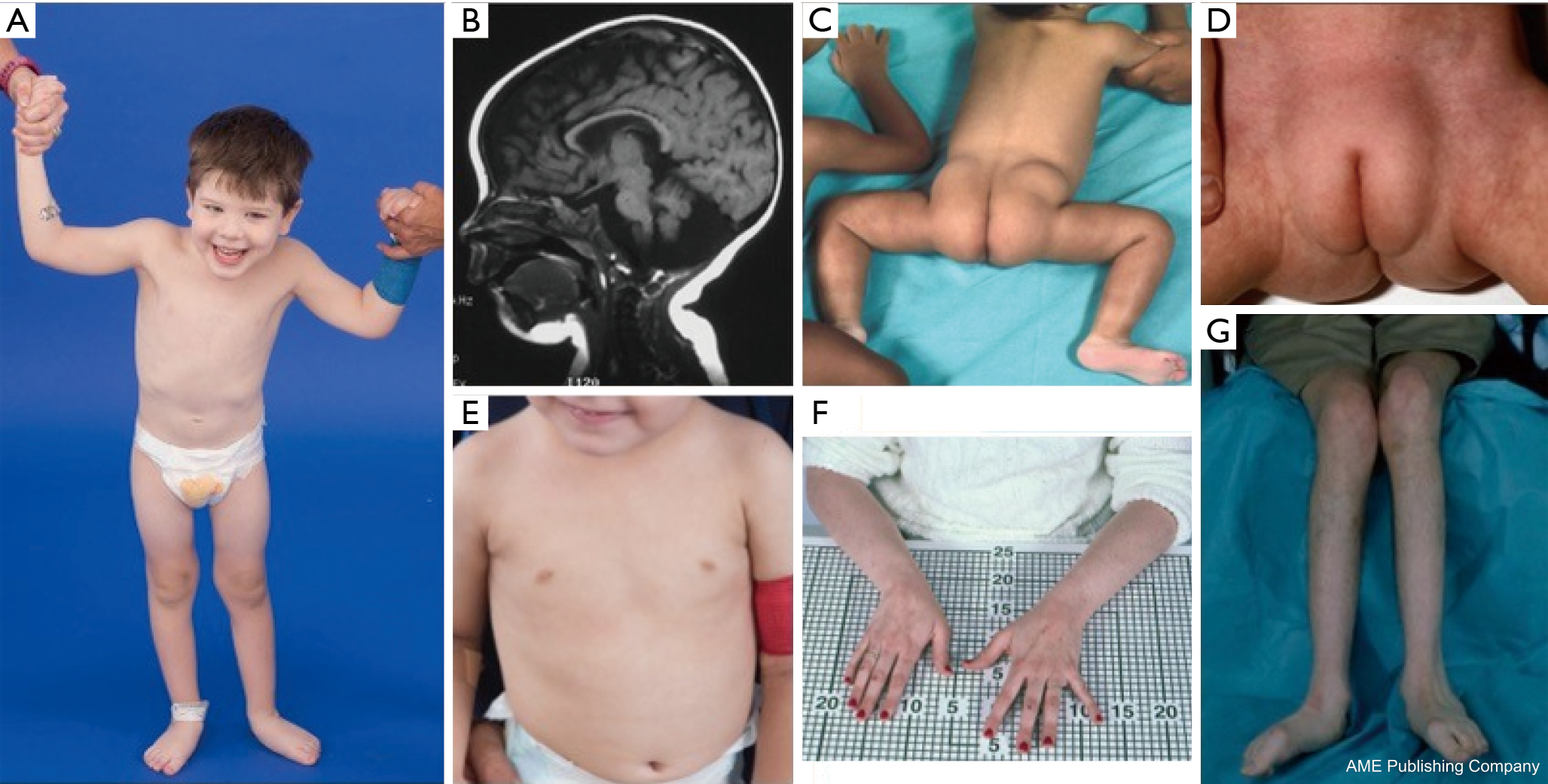
En la infancia, los individuos afectados pueden desarrollar retinitis pigmentosa, episodios similares a un ataque y convulsiones, retrasos en el habla y la motricidad, y neuropatía periférica. Desde el punto de vista constitucional, los pacientes suelen presentar un retraso en el crecimiento debido a anomalías alimentarias y gastrointestinales, así como un retraso global del desarrollo. Pueden observarse transaminasas hepáticas elevadas sin consecuencias clínicas, que suelen normalizarse a los 5 años de edad con fluctuaciones ocasionales con la enfermedad (21,24). Las biopsias hepáticas rara vez están indicadas en el CDG, a menos que se sospeche una fibrosis hepática (1). El hipotiroidismo clínico es raro, pero a los pacientes con CDG se les deben medir las hormonas tiroideas y la T4 libre, que pueden mostrar una globulina fijadora de tiroides (TBG) baja y elevaciones transitorias de la hormona estimulante del tiroides (TSH) (37). No se han descrito malformaciones del hígado ni de los conductos biliares en pacientes con PMM2-CDG.
Los adultos con PMM2-CDG pueden vivir hasta la séptima u octava década con un retraso cognitivo estable, neuropatía periférica y cifosis torácica y espinal progresiva con osteopenia u osteoporosis (34). La ataxia cerebelosa es un síntoma cada vez más reconocido junto con la afectación multisistémica (38-40). Las anomalías endocrinas incluyen hiperprolactinemia, liberación de la hormona del crecimiento con hiperglucemia, resistencia a la insulina e hipoglucemia hiperinsulinémica (41,42). En las mujeres afectadas, el hipogonadismo hipogonadotrópico puede conducir a la ausencia de desarrollo sexual secundario o a la ausencia de ovarios (41,43,44). Los pacientes pueden tener un mayor riesgo de trombosis debido a la disminución de los factores de coagulación séricos, incluidos los factores IV, IX y XI, la antitrombina III, la proteína C y la proteína S (29).
MPI-CDG (CDG-Ib, deficiencia de manosa-fosfato isomerasa)
MPI-CDG es única porque los pacientes afectados tienen poca o ninguna afectación neurológica y algunas manifestaciones de la enfermedad son tratables con manosa oral (2). Los síntomas son principalmente hepático-intestinales sin rasgos dismórficos ni retrasos cognitivos. Los pacientes suelen presentar vómitos recurrentes, hipoglucemia significativa, retraso en el crecimiento, enteropatía por pérdida de proteínas potencialmente mortal, cambios fibróticos en el hígado y dilatación de los conductos biliares (45-51). Los pacientes tienen un mayor riesgo de sufrir eventos trombóticos debido a las bajas concentraciones séricas de proteína C y S, y de antitrombina III.
ALG6-CDG (deficiencia de glucosiltransferasa 1)
ALG6-CDG es el segundo defecto de N-glicosilación más común caracterizado por un fenotipo similar pero más leve que PMM2-CDG. Los pacientes con ALG6-CDG presentan retraso en el desarrollo, hipotonía, convulsiones, estrabismo, ataxia, coagulopatía y dismorfismos faciales (es decir, orejas de implantación baja, hipertelorismo y macroglosia). Al igual que la MPI-CDG, también pueden presentar enteropatía con pérdida de proteínas. Además, los pacientes afectados pueden presentar anomalías esqueléticas, como braquidactilia, malformaciones en los dedos y escoliosis. Los pacientes afectados no suelen tener retinitis pigmentaria ni hipoplasia cerebelosa (52).
Defectos de glicosilación ligados a O y defectos combinados de glicosilación ligados a N y O
Debido a la presencia sustancial de glicanos O en las proteínas que contienen mucina, incluidos los glicosaminoglicanos (GAG) y las superficies epiteliales (53), los trastornos de la síntesis de GAG suelen dar lugar a displasias esqueléticas o enfermedades del tejido conectivo. Los pacientes afectados pueden presentar anomalías musculoesqueléticas, cutáneas y articulares (por ejemplo, laxitud articular, exostosis múltiples, condro/osteosarcomas) además de síntomas neurológicos (54-56). Por ejemplo, la N-acetilgalactosaminiltransferasa 3 (GALNT3) O-glicosila la hormona fosfatúrica, FGF23, impidiendo la escisión proteolítica y permitiendo su secreción intacta. La deficiencia de GALNT3 conduce a la calcinosis tumoral familiar, caracterizada por hiperfosfatemia y calcificaciones ectópicas (57,58).
Defectos de glicosilación de lípidos y de biosíntesis de anclajes GPI
Los glicoesfingolípidos y sus derivados sialilados, los gangliósidos, se expresan principalmente en las neuronas. Los defectos en la descomposición de los gangliósidos conducen a la acumulación y a las bien caracterizadas enfermedades de almacenamiento lisosomal. En el extremo opuesto, los defectos en la biosíntesis de gangliósidos, como ST3GAL5-CDG y B4GALNT1-CDG, son extremadamente raros y conducen a enfermedades neurodegenerativas graves. Los pacientes pueden presentar paraplejia espástica, retraso intelectual grave, epilepsia y síntomas no neurológicos que incluyen displasia esquelética, rasgos dismórficos y pigmentación anormal de la piel (59,60).
Las mutaciones en muchos genes dentro de la vía de biosíntesis del ancla GPI causan una variedad de anomalías congénitas múltiples, discapacidad intelectual y epilepsia. El defecto de biosíntesis de GPI mejor caracterizado, la deficiencia de PIGA ligada al X, se presenta con espasmos infantiles con hipsarritmia, hipotonía, múltiples anomalías cerebrales y dismorfismos faciales. Los pacientes también pueden presentar enfermedades cutáneas, hepáticas, cardíacas y renales variables (61-69). Algunas mutaciones dentro de PIGA causan la enfermedad fenotípicamente distinta hemoglobinuria paroxística nocturna (HPN), un trastorno adquirido de insuficiencia de la médula ósea (70,71).
Diagnóstico
Cuando se sospecha clínicamente de una CDG, el primer paso es solicitar pruebas bioquímicas de CDG en plasma o suero, incluyendo pruebas de CDT y N-glicanos. Los análisis de CDT y N-glicanos en suero sólo pueden detectar defectos de N-glicosilación, por lo que no serían útiles para diferenciar defectos aislados de O-glicosilación o anclaje de GPI. El análisis de las isoformas de transferrina se obtuvo originalmente mediante el enfoque isoeléctrico de la transferrina, ya que el fallo en la síntesis de N-glicanos provoca una deficiencia parcial de ácido siálico, que altera la carga de la transferrina sérica y posteriormente su migración catódica en un campo electroforético. Sin embargo, los análisis basados en la espectrometría de masas de la transferrina y el N-glicano han sustituido ahora en gran medida el enfoque isoeléctrico al identificar cambios específicos en los oligosacáridos por su masa y carga (72).
Defectos de glicosilación de proteínas ligadas a N
Los resultados del CDT de transferrina en suero se informan como la relación de transferrina mono-oligosacárido/di-oligosacárido, a-oligosacárido/di-oligosacárido transferrina, tri-sialo/di-oligosacárido transferrina, apolipoproteína CIII-1/apolipoproteína CIII-2, y ratio apolipoproteína CIII-0/apolipoproteína CIII-2. Estos resultados cuantitativos también vendrán acompañados de una interpretación del patrón de hallazgos.
Un patrón tipo I de CDT de transferrina se caracteriza por un aumento de las bandas de di- y asialotransferrina, e indica defectos en la síntesis de N-glicanos en el citosol o el retículo endoplásmico. Un patrón de tipo II se caracteriza por un aumento de las bandas de di- y asialotransferrina, y de las bandas de tri- y/o monosialotransferrina, e indica defectos en el procesamiento de N-glicanos en el aparato de Golgi (73).
Si se detecta un patrón de CDT de transferrina en suero de tipo I, la deficiencia de PMM2 o la de MPI deben estar a la cabeza de los diagnósticos diferenciales, ya que la PMM2-CDG es la CDG más común y la MPI-CDG es tratable y potencialmente mortal si no se trata. Para diferenciar los diagnósticos, deben realizarse perfiles de N-glicanos, secuencias moleculares o pruebas enzimáticas. El diagnóstico de PMM2-CDG o MPI-CDG se confirma mediante pruebas moleculares que muestran variantes patogénicas bialélicas en PMM2 o MPI, seguidas de la actividad enzimática de PMM o MPI en leucocitos o fibroblastos si la patogenicidad de las variantes genéticas es incierta. El análisis de N-glicanos o el análisis molecular diferenciarían la mayoría de ALG-CDG de PMM2 o MPI-CDG (15).
Un patrón de CDT de transferrina sérica tipo II indica defectos de Golgi como la deficiencia de N-acetilglucosaminiltransferasa (GnT) II (CDG tipo IIA, MGAT2-CDG). El análisis de la isoforma de la apolipoproteína CIII (Apo-CIII) es una prueba complementaria para un perfil CDT de tipo II, ya que mide los defectos de glicosilación de la mucina de tipo O en el aparato de Golgi. La sensibilidad de la CDT o de la Apo-CIII para detectar la CDG de tipo II es limitada. Por lo tanto, el perfil de N-glicanos y O-glicanos y el panel molecular o la secuenciación del exoma deben llevarse a cabo cuando estas pruebas clínicas estén disponibles. Los patrones de glicosilación de la transferrina pueden normalizarse esporádicamente; por lo tanto, puede estar indicado repetir las pruebas en pacientes con un alto índice de sospecha. Pueden obtenerse falsos positivos en pacientes con crisis agudas de intolerancia hereditaria a la fructosa, galactosemia, enfermedad hepática aguda y algunas infecciones bacterianas. Ninguna de las pruebas bioquímicas de CDG puede detectar todos los CDG, por lo que, incluso en presencia de resultados de cribado normales, pueden realizarse pruebas de panel de genes moleculares o secuenciación del exoma en caso de fuerte sospecha clínica. A la inversa, la confirmación bioquímica y funcional de los hallazgos genéticos moleculares también es esencial, ya que la mayoría de los pacientes con CDG son portadores de al menos una mutación de sentido erróneo leve y, a menudo, novedosa.
Defectos de glicosilación ligados a O y defectos de glicosilación combinados ligados a N y O
El diagnóstico se basa en la secuenciación molecular, ya que el análisis de la isoforma de transferrina no detectaría defectos de glicosilación O aislados. Los defectos combinados de glicosilación ligados a N y O pueden detectarse mediante CDT, análisis de ApoCIII y análisis de N-glicanos y O-glicanos en plasma.
Defectos de glicosilación de lípidos y de biosíntesis de anclajes GPI
La citometría de flujo de los granulocitos sanguíneos mide la expresión de la superficie celular de las proteínas ancladas a GPI, como CD16 y CD24. El análisis por citometría de flujo de los glóbulos blancos o rojos para determinadas proteínas de la superficie celular ancladas a la GPI está disponible clínicamente como prueba de la HPN debido a mutaciones adquiridas en el gen PIGA. La prueba de la HPN puede revelar anomalías en otras deficiencias de anclaje GPI, pero el diagnóstico se basa principalmente en el análisis molecular.
Análisis molecular
El mayor rendimiento diagnóstico para la HPN es el panel de secuenciación genética basado en la próxima generación o la secuenciación clínica del exoma (CES). La secuenciación genética revisa la secuencia de nucleótidos, o la ortografía de las «letras», de los genes para determinar si hay un cambio que afecte a la función del gen. El genoma humano consta de 3 millones de nucleótidos, pero sólo el 1-2% de ellos, llamados exones, se traducen en un producto proteico funcional. El resto del ADN no codificante intercalado entre los exones que no se traduce se llama intrones (74). La CES examina casi todos los exones conocidos de los aproximadamente 20.000 genes del genoma humano, que representan una minoría del material genético de los cromosomas, pero que tienen más probabilidades de contener variantes causantes de enfermedades (patógenas). La CES también puede incluir la secuenciación del ADN mitocondrial (ADNmt), que interroga al pequeño ADN circular extranuclear localizado en las mitocondrias que se hereda exclusivamente por vía materna.
Los posibles resultados de la CES incluyen resultados positivos, negativos y variantes de significado desconocido. Un resultado positivo significa que se identifican variantes conocidas causantes de la enfermedad (es decir, patógenas), tras lo cual se puede discutir el diagnóstico, la historia natural, el pronóstico, el riesgo de recurrencia y las opciones de tratamiento. Un resultado negativo significa que no se han identificado variantes patogénicas detectables. Las variantes de significado desconocido (también conocidas como VUS) significan que, aunque se han identificado cambios genéticos, no hay suficiente información sobre el cambio genético específico para saber definitivamente si es causante de la enfermedad. Es de esperar que haya variaciones en el ADN de cada individuo, por lo que el hecho de que las muestras de los padres se analicen simultáneamente para compararlas puede ayudar a la interpretación de los resultados en el laboratorio y en la clínica. El rendimiento diagnóstico de la CES se estima en un 30-35% y está aumentando con el tiempo a medida que el descubrimiento de genes y el conocimiento del genoma humano siguen progresando (75-77). La CES se solicita cada vez más como prueba genética amplia de primera línea, dado su rápido tiempo de respuesta y su bajo coste relativo para la cantidad de información genética analizada. Las limitaciones de la CES incluyen la falta de una sensibilidad del 100%, la incapacidad de detectar ciertos tipos de cambios genéticos (por ejemplo, deleciones, duplicaciones, repeticiones de trinucleótidos, mutaciones intrónicas profundas o defectos de metilación), y el hecho de que un diagnóstico puede no proporcionar información adicional sobre la enfermedad o cambiar el manejo.
En el informe de la CES, las variantes patogénicas detectadas incidentalmente en genes asociados con condiciones genéticas bien conocidas pueden ser reportadas como hallazgos secundarios (78). Esta lista de enfermedades recomendadas está elaborada por el American College of Medical Genetics (ACMG). La Ley de No Discriminación de la Información Genética (GINA, por sus siglas en inglés) es una consideración importante a la hora de decidir si se acepta o no el aprendizaje de los hallazgos incidentales (79). La GINA protege a las personas contra el uso indebido de la información genética en los seguros médicos y el empleo, pero no en los seguros de vida. La GINA protege la siguiente información genética: historial médico familiar, pruebas de portadores, pruebas genéticas prenatales, pruebas de susceptibilidad y predictivas, y análisis de tumores u otras evaluaciones de genes, mutaciones o cambios cromosómicos.
Manejo
El manejo del CDG depende en gran medida de los síntomas específicos del individuo. Los síntomas recurrentes en los pacientes con CDG incluyen retraso en el crecimiento, retraso global en el desarrollo, vómitos, episodios similares a un derrame cerebral y anomalías esqueléticas. También suelen observarse coagulopatías clínicas o subclínicas, endocrinopatías, hepatopatías y defectos cardíacos. Se recomiendan pruebas de laboratorio de referencia para establecer el alcance de la enfermedad y un seguimiento rutinario, especialmente para la PMM2-CDG. Éstas incluyen pruebas de la función hepática, albúmina sérica, pruebas de la función tiroidea, incluida la T4 libre, proteína C, proteína S, antitrombina III, factor IX, análisis de orina y gonadotropinas séricas y hormona del crecimiento.
Las pruebas de imagen recomendadas incluyen ecocardiograma, ecografía renal, edad ósea, examen oftalmológico para evaluar el cristalino, la retina, la movilidad ocular y la presión intraocular. A menos que se indique lo contrario, se recomiendan las vacunas rutinarias para los adultos y los niños afectados por CDG. Deben obtenerse títulos de anticuerpos después de la vacunación, ya que los pacientes pueden tener una respuesta inmunógena subóptima. Puede ser necesaria la repleción profiláctica de los factores de coagulación antes de cualquier procedimiento quirúrgico si existen deficiencias al inicio.
Debe realizarse una evaluación genética clínica para discutir los aspectos hereditarios de la CDG, así como establecer un hogar médico para estos complejos pacientes. El hogar médico suele ser el servicio de genética bioquímica, aunque los departamentos de genética, neurodesarrollo o neurología también han desempeñado esta función si no se dispone de un servicio dedicado a la genética bioquímica. A menudo es necesaria la derivación a especialistas en gastroenterología, hematología, endocrinología, apoyo nutricional, terapias del habla, ocupacionales, físicas y de alimentación, ortopedia y medicina de rehabilitación.
Terapias dirigidas y pronóstico
El tratamiento para la mayoría de los tipos de CDG es en gran medida de apoyo, con algunas excepciones. La CDG-PMI es la más eficazmente tratable de todas las CDG. La manosa oral es convertida en manosa-6-fosfato por las hexocinasas intracelulares, con lo que se evita el bloqueo enzimático y se produce el sustrato deficiente. La administración de suplementos de manosa suele comenzar con 1 g/kg de peso corporal al día, dividido en 4-6 dosis al día. Mientras que la enteropatía por pérdida de proteínas, potencialmente mortal, responde especialmente al tratamiento con manosa, la enfermedad hepática en la MPI-CDG puede seguir progresando. Los síntomas clínicos mejoran rápidamente y la CDT de transferrina se normaliza a lo largo de los meses, aunque la enfermedad hepática puede seguir progresando con el tratamiento (45,80,81).
Hay que tener precaución con la administración de suplementos de manosa durante el embarazo, ya que la administración de manosa en modelos de ratones hipomórficos de fosfomanosa isomerasa preñados provocó letalidad embrionaria y ceguera en sus crías (82). Además, la manosa intravenosa se ha asociado con una disminución de la conciencia y convulsiones, que se resolvieron con la administración de glucosa (83).
El tratamiento de la PMM2-CDG es en gran medida de apoyo y se basa en la sintomatología. Sin embargo, actualmente se están desarrollando próximos ensayos clínicos sobre la terapia de sustitución del sustrato manosa-1-fosfato.
Para otros CDG, se han investigado varios azúcares simples orales con el objetivo de mejorar teóricamente la hipoglucosilación. Se ha probado la fucosa para el SLC35C1-CDG y la galactosa para el PGM1-CDG y el SLC35A2-CDG con resultados mixtos (84). Se ha demostrado que la D-galactosa a 1,0-2,5 g/kg/día (máximo 50 gramos) mejora la hipoglucemia, la coagulopatía y la endocrinopatía en PGM1-CDG (85,86). También se ha demostrado que la galactosa mejora la endocrinopatía y la coagulopatía en TMEM165-CDG (87) y SLC39A8-CDG. También se informó de una mejora clínica considerable en pacientes con SLC39A8-CDG con 15-20 mg/kg/día de MnSO4 (88). Se están realizando ensayos clínicos para investigar la utilidad de la N-acetilmanosamina (ManNAc) en la GNE-CDG (89), y se están realizando varios ensayos preclínicos para otras CDG (90).
A pesar de los avances médicos, existe una mortalidad significativa en los niños con CDG durante el primer año de vida por fallo multiorgánico o infección grave (91). Los niños con CDG pueden presentar una enfermedad multiorgánica fulminante, convulsiones intratables o hipoalbuminemia grave que progresa a anasarca. Algunos pacientes responden a la diuresis agresiva y a la reposición de albúmina, mientras que otros son refractarios al tratamiento. Se ha demostrado que el butirato de sodio mejora el control de las convulsiones en la CAD-CDG y la PIGM-CDG (92). También se ha demostrado que la dieta cetogénica disminuye la frecuencia de las convulsiones en algunos casos de PIGA-CDG (93). Durante los episodios similares a un ictus, la hidratación intravenosa y el mantenimiento de una glucemia normal pueden ser útiles mientras se descarta la etiología trombótica o hemorrágica vascular subyacente.
Con la llegada de las técnicas de edición del genoma y una mejor comprensión del mecanismo de las enfermedades englobadas en el paraguas diagnóstico del CDG, el futuro del desarrollo terapéutico dirigido sigue siendo prometedor.
Agradecimientos
Nos gustaría dar las gracias a Lynne Wolfe, ARNP y a Donna Krasnewich, MD, PhD por proporcionarnos las fotos clínicas obtenidas como parte de las Investigaciones Clínicas y Básicas en Trastornos Congénitos de Glicosilación Conocidos y Sospechados (NCT02089789). También nos gustaría agradecer a Jenny Thies, MS, LGC por su experiencia en el asesoramiento genético.
Financiación: IJ Chang cuenta con el apoyo de los Institutos Nacionales de Salud T32GM007454.
Nota
Conflictos de intereses: Los autores no tienen conflictos de intereses que declarar.
Consentimiento informado: Se obtuvo el consentimiento informado por escrito de los pacientes para la publicación de este manuscrito y cualquier imagen que lo acompañe.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutaciones en PMM2 que causan trastornos congénitos de la glicosilación, tipo Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. La prevalencia de PMM2-CDG en Estonia basada en las frecuencias de portadores de la población y los pacientes diagnosticados. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Falta de equilibrio Hardy-Weinberg para la mutación PMM2 más prevalente en CDG-Ia (trastornos congénitos de la glicosilación tipo Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;144:245-7. ¡
- Jaeken J, Hennet T, Matthijs G, et al. CDG nomenclature: time for a change! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. ¿Qué hay de nuevo en CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Trastornos congénitos de la glicosilación. Parte I. Defectos de la N-glicosilación de las proteínas. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Hallazgos clínicos, de laboratorio y moleculares y datos de seguimiento a largo plazo en 96 pacientes franceses con PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) y revisión de la literatura. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. Una encuesta nacional de PMM2-CDG en Italia: alta frecuencia de una variante neurológica leve asociada a la mutación L32R. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG in South Africa: Descripción del genotipo-fenotipo de cinco nuevos pacientes. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Different glycosyltransferases are differentially processed for secretion, dimerization, and autoglycosylation. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. En: Mora-Montes H. Editor. Glycans: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Skin manifestations in CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. Sistema IGF en niños con trastornos congénitos de la glicosilación. Clin Endocrinol (Oxf) 2009;70:892-7.
- De Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertal growth in congenital disorder of glycosylation type Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N-glycan synthesis. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–una enfermedad metabólica hereditaria recientemente descubierta. Múltiples manifestaciones orgánicas, incidencia 1/80.000, difícil de tratar. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Grave isquemia miocárdica transitoria causada por miocardiopatía hipertrófica en un paciente con trastorno congénito de la glicosilación tipo Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Características clínicas en adultos con trastornos congénitos de la glicosilación tipo Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Síndrome de glicoproteína deficiente en carbohidratos. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. La modificación de O-GlcNAc de la proteína de la cubierta del potyvirus Plum pox virus mejora la infección viral. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Scandinavian CDG-Ia patients: genotype/phenotype correlation and geographic origin of founder mutations. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 pacientes adultos franceses con trastorno congénito de la glicosilación PMM2: resultado del fenotipo pediátrico clásico y descripción de un fenotipo de inicio tardío. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Trastorno congénito de la glicosilación tipo Ia (CDG-Ia): espectro fenotípico del genotipo R141H/F119L. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Thyroid function in PMM2-CDG: diagnostic approach and proposed management. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Nuevos trastornos en el metabolismo de los carbohidratos: trastornos congénitos de la glicosilación y su impacto en el sistema endocrino. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- De Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadal function and glycoprotein hormones in the carbohydrate-deficient glycoprotein (CDG) syndrome. Acta Paediatr 1995;84:655-9.
- De Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Complicaciones cardíacas de los trastornos congénitos de la glicosilación (CDG): una revisión sistemática de la literatura. J Inherit Metab Dis 2017;40:657-72.
- De Koning TJ, Toet M, Dorland L, et al. Hidropesía fetal no inmune recurrente asociada al síndrome de glicoproteína deficiente en carbohidratos. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Deficiencia de fosfomanosa isomerasa y terapia con manosa. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- De Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: Una nueva manifestación del síndrome de glicoproteína deficiente en carbohidratos tratable con manosa. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG6 trastorno congénito defectuoso de la glicosilación. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: De la producción a la secreción. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Resultado clínico a largo plazo y variabilidad fenotípica en la calcinosis tumoral familiar hiperfosfatémica y el síndrome de hiperostosis hiperfosfatémica causado por una nueva mutación GALNT3; informe de un caso y revisión de la literatura. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. El heparán sulfato en el desarrollo, crecimiento y patología del esqueleto: el caso de las exostosis múltiples hereditarias. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Afecciones musculoesqueléticas no inflamatorias diversas. Calcinosis tumoral familiar hiperfosfatémica (FGF23, GALNT3 y αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. La ablación del gen Galnt3 conduce a bajas concentraciones intactas de factor de crecimiento de fibroblastos 23 (Fgf23) en circulación y a hiperfosfatemia a pesar de una mayor expresión de Fgf23. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Una mutación en una enzima biosintética de gangliósidos, ST3GAL5, resulta en el síndrome de la sal &pimienta, un trastorno neurocutáneo con alteración de la glicolipidación y glicoproteína. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutaciones en B4GALNT1 (GM2 sintasa) subyacen a un nuevo trastorno de la biosíntesis de gangliósidos. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. Mutaciones en PIGA causan encefalopatías epilépticas de inicio temprano y características distintivas. Neurology. Neurología 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Mutaciones hipomórficas en PGAP2, que codifica una proteína de remodelación de anclaje GPI, causan discapacidad intelectual autosómica-recesiva. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. El fenotipo de una mutación de línea germinal en PIGA: el gen somáticamente mutado en la hemoglobinuria paroxística nocturna. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutaciones en PIGO, un miembro de la vía de síntesis de anclaje GPI, causan hiperfosfatasia con retraso mental. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. Un nuevo síndrome de discapacidad intelectual causado por la deficiencia de anclaje GPI debido a mutaciones homocigóticas en PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutaciones en el gen del glucosilfosfatidilinositol PIGL causan el síndrome CHIME. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. La hemoglobinuria paroxística nocturna (PNH) está causada por mutaciones somáticas en el gen PIG-A. EMBO J 1994;13:110-7.
- Brodsky RA. Hemoglobinuria paroxística nocturna. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distribuciones de exones e intrones en el genoma humano. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Secuenciación clínica del exoma completo para el diagnóstico de trastornos mendelianos. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. La mejora del rendimiento diagnóstico en comparación con los paneles de secuenciación de genes dirigidos sugiere un papel para la secuenciación del genoma completo como prueba genética de primer nivel. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. El costo y el rendimiento diagnóstico de la secuenciación del exoma para niños con sospecha de trastornos genéticos: un estudio de referencia. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recomendaciones para informar de los hallazgos secundarios en la secuenciación clínica del exoma y del genoma, actualización de 2016 (ACMG SF v2.0): una declaración de política del Colegio Americano de Genética Médica y Genómica. Genet Med 2017;19:249-55.
- Rothstein MA. Corrientes en la ética contemporánea. GINA, la ADA y la discriminación genética en el empleo. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Tromboembolismo recurrente en un niño con un trastorno congénito de la glicosilación (CDG) tipo Ib y tratamiento con manosa. Pediatr Hematol Oncol 2008;25:762-8.
- Mención K, Lacaille F, Valayannopoulos V, et al. Desarrollo de enfermedad hepática a pesar del tratamiento con manosa en dos pacientes con CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Los suplementos de manosa inducen letalidad embrionaria y ceguera en ratones hipomórficos de fosfomanosa isomerasa. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Seizures and stupor during intravenous mannose therapy in a patient with CDG syndrome type 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Suplemento de fucosa en la deficiencia de adhesión leucocitaria tipo II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Inherited glycosylphosphatidyl inositol deficiency: a treatable CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Suplementación oral de D-galactosa en PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. La suplementación con galactosa en pacientes con TMEM165-CDG rescata los defectos de glicosilación. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. Deficiencia de SLC39A8: corrección bioquímica y mejora clínica importante mediante terapia de manganeso. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Terapias orales de monosacáridos para revertir la hiposialilación renal y muscular en un modelo de ratón de miopatía GNE. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Human glycosylation disorders. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Dieta cetogénica – Un nuevo tratamiento para la encefalopatía epiléptica temprana debido a la deficiencia de PIGA. Brain Dev 2016;38:848-51.