- Overblik
- Epidemiologi
- Biokemisk klassifikation og nomenklatur
- Genetik
- Patofysiologi
- N-linkede proteinglykosyleringsdefekter
- O-linkede glykosyleringsdefekter og kombinerede N- og O-linkede glykosyleringsdefekter
- Lipidglykosylering og GPI-ankerbiosyntesedefekter
- Kliniske manifestationer
- N-linkede proteinglykosyleringsdefekter
- PMM2-CDG (CDG-Ia, PMM2-mangel)
- MPI-CDG (CDG-Ib, mannosephosphat isomerase deficiency)
- ALG6-CDG (glucosyltransferase 1-mangel)
- O-linkede glykosyleringsdefekter og kombinerede N- og O-linkede glykosyleringsdefekter
- Defekter i glykosylering af lipider og GPI-ankerbiosyntese
- Diagnose
- N-bundne proteinglykosyleringsdefekter i proteiner
- O-linkede glykosyleringsdefekter og kombinerede N- og O-linkede glykosyleringsdefekter
- Lipidglykosylerings- og GPI-ankerbiosyntesedefekter
- Molekylær analyse
- Behandling
- Targeted terapier og prognose
- Anerkendelser
- Fodnote
Overblik
Glykosylering er processen med at tilføje sukkerrester til proteiner og lipider i forskellige cellulære veje. Medfødte glykosyleringsforstyrrelser (CDG) er en genetisk og klinisk heterogen gruppe på over hundrede sygdomme, der skyldes defekter i forskellige trin langs glykansyntesen eller modifikationsvejene. De fleste af disse monogene sygdomme nedarves autosomalt recessivt, men der er også beskrevet autosomalt dominerende og X-bundne former.
CDG viser sig typisk med multisystemiske manifestationer, som oftest udviklingsforsinkelse, manglende trivsel, hypotoni, neurologiske abnormiteter, hepatopati og koagulopati. De ramte personer kan også have øjen-, hud- og hjertesygdomme samt dysmorfismer i ansigtet. Selv om neurologiske ændringer og kognitive forsinkelser ses hos størstedelen af de berørte personer, er der visse tilfælde og endda typer, som ikke har neurologiske manifestationer. I betragtning af den brede kliniske og genetiske ætiologi af CDG er den kliniske diagnose afhængig af et højt mistænksomhedsindeks ved multisystemisk sygdom.
Serum carbohydratdeficient transferrin (CDT) analyse er den første screeningtest hos patienter med mistanke om CDG, men er begrænset i påvisning til N-glykosyleringsdefekter med sialinsyremangel. Næste test omfatter dolichol-linked glycan-analyse og genetisk testning. Det er vigtigt at stille tidlig diagnose af denne gruppe af eksponentielt voksende sygdomme, da nogle CDG’er kan behandles. Behandlingen af glykosyleringsdefekter er hovedsagelig understøttende, selv om der findes målrettede behandlinger for MPI-CDG, SLC35C1-CDG, PIGM-CDG og PGM1-CDG. Nærmere oplysninger om disse behandlinger findes under afsnittet “Målrettede terapier og prognose” nedenfor. Fokus i denne gennemgang vil være på de mest almindelige typer af CDG med genkendelige fænotyper eller behandlinger, og målgruppen er leverandører af primær sundhedspleje.
Epidemiologi
Incidensen og prævalensen af alle typer af CDG samlet set er ikke veletableret, selv om der er rapporteret om patienter verden over fra næsten alle etniske baggrunde, og begge køn er lige hårdt ramt. Den anslåede prævalens i europæiske og afroamerikanske befolkninger er 1/10.000 baseret på bærerfrekvenser af kendte patogene varianter i 53 gener (1-4). Prævalensen af den mest almindeligt diagnosticerede CDG, PMM2-CDG, varierer fra 1/20.000 i hollandske befolkninger og 1/77.000 i Estland på grundlag af isolerede rapporter (5,6). Hidtil er der rapporteret færre end 100 tilfælde for de fleste CDG-typer.
Biokemisk klassifikation og nomenklatur
Bredt set klassificeres CDG i øjeblikket i fire kategorier – (I) N-linked glykosylering, (II) O-linked glykosylering, (III) kombineret N- og O-linked/multipel glykosylering og (IV) lipid- og glykosylphosphatidylinositol (GPI) ankerbiosyntesedefekter.
Den N-bundne proteinglykosyleringsdefekt PMM2-CDG, tidligere kendt som CDG type Ia, var den første CDG, der blev rapporteret af Jaeken i 1980, og den er stadig langt den mest almindelige CDG til dato (7). PMM2-CDG blev oprindeligt kaldt “carbohydratdeficient glycoproteinsyndrom” på grund af de mange serumglycoproteinafvigelser, der blev set ved isoelektrisk fokusering af serumtransferrin hos de berørte personer. Historisk set blev CDG klassificeret efter mønstre af transferrin-isoformanalyse – type I-mønstre blev tilskrevet dolichol-bundne glykan-samlings- og overførselsdefekter lokaliseret til cytoplasmaet eller ER, og type II-mønstre blev tilskrevet forarbejdningsdefekter i Golgi-apparatet. Fra dette forgreningssted blev CDG’er derefter navngivet alfabetisk i den rækkefølge, de blev opdaget.
Med fremkomsten af udbredt molekylær diagnostik blev CDG-nomenklaturen opdateret i 2008 for at specificere den molekylære sygdomsætiologi, hvilket afspejler den eksponentielle vækst af sygdomsveje og lidelser, der ikke passede pænt ind i de tidligere etablerede dikotomiske kategorier. I øjeblikket angives CDG-nomenklaturen ved det berørte gennavn (ikke kursiv, gennavne på www.genenames.org) efterfulgt af -CDG (f.eks. PMM2-CDG) (8).
Genetik
Den store majoritet af medfødte glykosyleringsforstyrrelser nedarves autosomalt recessivt, hvor en mutation nedarves fra hver asymptomatisk (bærer) forælder. Molekylær testning, normalt med næste generations sekventeringsmetoder, er nødvendig for at stille en genetisk diagnose. Ved at teste for den kendte variant hos forældrene kan man bekræfte arvelighed versus de novo-forekomst. Ved autosomal recessiv arv er risikoen for gentagelse for søskende og hver graviditet hos et ramt individ 25 % for at være ramt, 50 % for at være asymptomatisk bærer og 25 % for at være upåvirket.
En håndfuld CDG har autosomal dominant arv (N-linked: GANAB-CDG, PRKCSH-CDG; O-linked: GANAB-CDG, PRKCSH-CDG; O-linked: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Færre er X-bundne (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). De fleste dominerende og nogle X-linked former af CDG skyldes de novo-mutationer. Specifikke sygdomme og gener er beskrevet nedenfor i afsnittet “patofysiologi”.
Mutationsdata for alle offentliggjorte gener for CDG er tilgængelige på Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Oplysninger om specifikke genvarianter er tilgængelige på Leiden Open Variation Database med integrerede in silico patogenicitetsværktøjer (http://www.lovd.nl/3.0/home). Kliniske synopser for specifikke gener kan findes på Online Mendelian Inheritance in Man (http://www.omim.org/) eller i et mere begrænset omfang på GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). I betragtning af det lille antal berørte patienter for de fleste CDG-subtyper er genotype-fænotype-korrelation vanskelig at fastslå.
Patofysiologi
Over 130 typer af CDG er til dato blevet rapporteret (9,10). I betragtning af den allestedsnærværende tilstedeværelse af glykosyleringsveje er CDG ekstremt forskellige i deres biokemiske patogenese. Talrige proteiner og lipider (dvs. sfingolipider og glykolipider) gennemgår glykosylering med monosaccharider og/eller oligosaccharider, samlet benævnt glykaner, i forskellige cellulære kompartmenter. Deres subcellulære placering er forskellig, men de fleste defekter forekommer i ER- eller Golgi-apparatet. Kliniske træk og genetisk ætiologi af de mest almindelige CDG efter vej er opsummeret i tabel S1.
Men blandt proteiner beskrives glykaner ved deres binding til polypeptidkæden-N-glykaner er knyttet til amidgruppen af asparagin (Asn), mens O-glykaner er knyttet til hydroxylgruppen af enten serin eller threonin. N-glycansyntesen kræver trinvis opbygning af nukleotidforbundne sukkerstoffer i cytosolen, samling i det endoplasmatiske retikulum og forarbejdning i Golgi-apparatet. I modsætning hertil kræver O-glykansyntese samling, men ingen forarbejdning, og derfor forekommer fejl i O-glykosylering overvejende i Golgi-apparatet.
N-linkede proteinglykosyleringsdefekter
N-glykosylering indebærer kovalent binding af kulhydratstrukturer til sidekædens amidgruppe af Asn-rester inden for et konsensus Asn-X-Ser/Thr-acceptorsted, translokation af substratpolypeptidet til det endoplasmatiske retikulum til remodellering og yderligere modifikation af N-glykan-kæden i Golgi-apparatet (11,12). Defekter et hvilket som helst sted langs syntesen, samlingen og forarbejdningsvejen kan føre til klinisk sygdom.
PMM2-CDG skyldes patogene varianter i phosphomannomutase 2 (PMM2)-genet, hvilket fører til mangel på PMM2-enzymet, der katalyserer den cytosoliske omdannelse af mannose-6-fosfat til mannose-1-fosfat i det andet trin af guanosindiphosphat (GDP)-mannosesyntesen. De fleste patienter har sammensatte heterozygote patogene missense-mutationer (www.lovd.nl/PMM2). Den mest almindelige tilbagevendende patogene variant p.Arg141His findes hos ca. 40 % af de berørte personer af europæisk afstamning, og p.Phe119Leu findes også hyppigt i Nordeuropa (1). Der er rapporteret om genotype-fænotype korrelationer for PMM2-CDG (3,13,14).
MPI-CDG er en autosomal recessiv lidelse forårsaget af patogene varianter i mannosephosphatisomerase (MPI)-genet, der fører til mangelfuld phosphomannoseisomerase (MPI). MPI katalyserer normalt det første trin i GDP-mannosesyntesen (dvs. omdannelsen af fruktose-6-fosfat til mannose-6-fosfat), men fruktose-6-fosfat ophobes ikke intracellulært, da det også kan metaboliseres af den glykolytiske vej. Selv om MPI-CDG biokemisk set ligner PMM2-CDG, forårsager MPI-CDG derfor ikke så betydelig neurologisk og multisystemisk involvering. CDT er også den foretrukne screeningtest for MPI-CDG, som viser et type 1-mønster. Diagnosen kan derefter bekræftes molekylært eller ved fibroblast/leukocyt MPI-aktivitet.
ALG6-CDG er en recessiv sygdom forårsaget af mutationer i ALG6, hvilket fører til unormal tilknytning af tre glukosemolekyler til dolichol-bundne mannoseintermediater og downstream hypoglykosylering af serumglykoproteiner (15).
O-linkede glykosyleringsdefekter og kombinerede N- og O-linkede glykosyleringsdefekter
O-glykosylering omfatter den trinvise tilføjelse af kulhydratkæder til serin-, threonin- og hydroxylysinrester af proteiner ved hjælp af glykosyltransferaser i Golgi-apparatet (16). Flere typer O-linkede glykaner er blevet associeret med sygdomme hos mennesker og er navngivet efter det første sukkerstof, der er knyttet til aminosyreresterne (17).
Lipidglykosylering og GPI-ankerbiosyntesedefekter
GPI-ankre er glykolipider, der undergår sekventiel samling i det endoplasmatiske retikulum og modifikationer i Golgi-anlægget. GPI-ankerbiosynteseforstyrrelser, der skyldes enzymmangler, er navngivet alfabetisk efter den rækkefølge, hvor de blev opdaget, og ikke kronologisk efter samlefasen. Når de er syntetiseret, sidder GPI-ankre på plasmamembraner og binder hundredvis af proteiner på celleoverfladen og udfører et væld af cellulære funktioner. De fleste af disse sygdomme er autosomalt recessive med den bemærkelsesværdige undtagelse af X-bundet PIGA-mangel.
Kliniske manifestationer
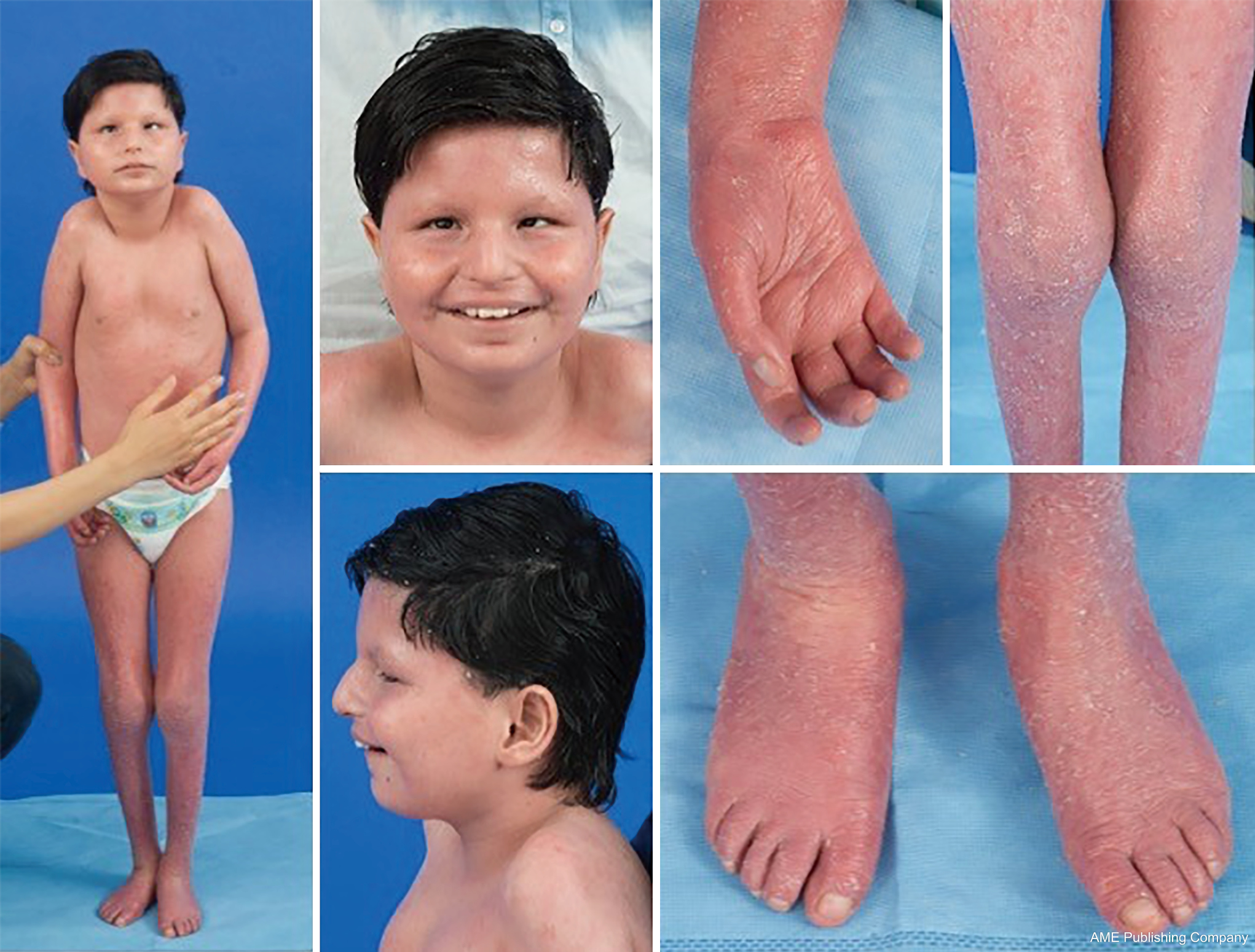
Givet den allestedsnærværende tilstedeværelse af glykosyleringsveje kan stort set ethvert organsystem være involveret i CDG, selv om de fleste tilfælde involverer neurologiske abnormiteter. Nogle CDG’er præsenterer sig med ichtyose, herunder MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (figur 1) og PIGL-CDG (18,19). Næsten alle CDG præsenterer multi-systemisk sygdom inden for de første par leveår, bortset fra at nogle kun påvirker et enkelt organsystem (f.eks, nethinden i DHDDS-CDG; det neuromuskulære knudepunkt i ALG2-CDG, ALG14-CDG, CFPT1-CDG; hjernen i ST3GAL3-CDG, TUSC3-CDG; huden eller skeletmuskulaturen i POGLUT1-CDG, POFUT1-CDG; brusk i EXT1/EXT2-CDG; leveren i TMEM199-CDG; røde blodlegemer i SEC23B-CDG). Debutalder og sværhedsgrad kan variere fra neonatal dødelig til næsten asymptomatisk voksenalder og enhver permutation derimellem. Den mest almindeligt rapporterede konstellation af symptomer omfatter udviklingsforsinkelse, manglende trivsel, hypotoni, neurologiske abnormiteter, hypoglykæmi og variable lever-, øjen-, hud-, gastrointestinale, immunologiske, skelet- og koagulationsanomalier (19).
Den fuldstændige fænotype for mange CDG-subtyper er endnu ikke fuldt ud afgrænset på grund af de sjældne rapporterede tilfælde. Derfor bør CDG overvejes i enhver situation med multisystemisk sygdom, især i tilfælde med en neurologisk komponent eller uspecifik udviklingsforsinkelse med uklar ætiologi.
Og selv om patofysiologien for multisymptomer stadig mangler at blive belyst, er forholdet mellem visse glykosyleringsveje og specifikke kliniske symptomer blevet afklaret. For eksempel skyldes manglende trivsel, der ses i mange typer CDG, hypoglykosylering og nedsat dannelse af flere glykoproteiner inden for insulinvækstvejen, herunder IGF-1, ALS og IGFBP-3 (20). Efterhånden som vi bedre forstår denne gruppe af komplekse lidelser, anerkendes CDG i stigende grad hos personer med undvigende diagnoser. Organsysteminddragelserne af forskellige CDG er opsummeret i tabel S1. Vi vil diskutere de kliniske træk ved de mest almindelige former og former med målrettede behandlinger af CDG nedenfor.
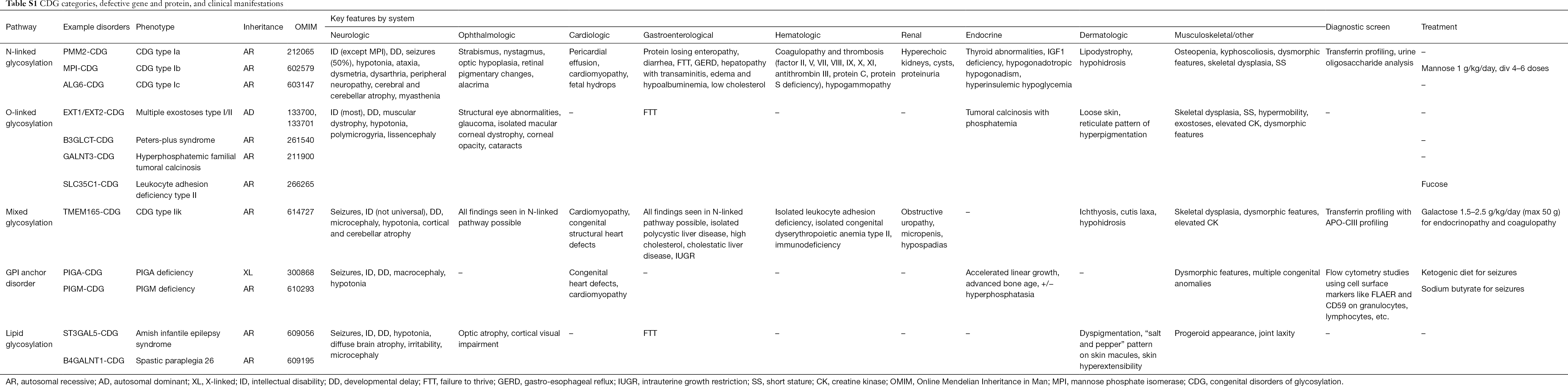
Fuldstændig tabel
N-linkede proteinglykosyleringsdefekter
Som den hyppigst diagnosticerede CDG bliver fænotypen af N-linkede glykosyleringsforstyrrelser ofte udråbt som den klassiske præsentation. Det fænotypiske spektrum af CDG er imidlertid ret forskelligartet, og mange CDG’er har måske ikke de stereotype symptomer, der er forbundet med PMM2-CDG.
PMM2-CDG (CDG-Ia, PMM2-mangel)
PMM2-CDG er den mest almindelige CDG med over 700 rapporterede tilfælde på verdensplan. Den er karakteriseret ved multisystemisk alvorlig sygdom i spædbarnsalderen, neurologisk sygdom og udviklingsforsinkelse i barndommen og/eller stabilt intellektuelt handicap i voksenalderen (21,22).
I spædbarnsalderen viser PMM2-CDG sig med neurologiske abnormiteter typisk kort efter fødslen, nemlig strabismus og unormale øjenbevægelser, cerebellær hypoplasi, hypotoni, psykomotorisk retardering, ataksi, hypotoni og hyporefleksi. Spædbørn kan også have leversygdom, nefrotisk syndrom og nyrecyster, perikardial effusion og hypertrofisk kardiomyopati, manglende trivsel og multiorgansvigt, der resulterer i død inden for det første leveår hos op til 20 % af de berørte individer (21,23-28).
En konstellation af dysmorfiske træk er blevet beskrevet hos patienter med PMM2-CDG (figur 2,3). Disse omfatter en hypoplastisk lillehjernen, ansigtsdysmorfismer (dvs. store, dysplastiske ører), inverterede brystvorter og unormal fordeling af fedtvæv over balderne eller suprapubisk region, som kan forsvinde med alderen (14,21,29-32). Patienterne er blevet beskrevet at have en udadvendt og glad opførsel. Præsentationen er meget variabel, selv om strabismus kan ses hos mere end 70 % af de berørte patienter (21,23,33-35). Inverterede brystvorter og unormale fedtpuder ses hos ca. 25-50 % af patienterne (36).

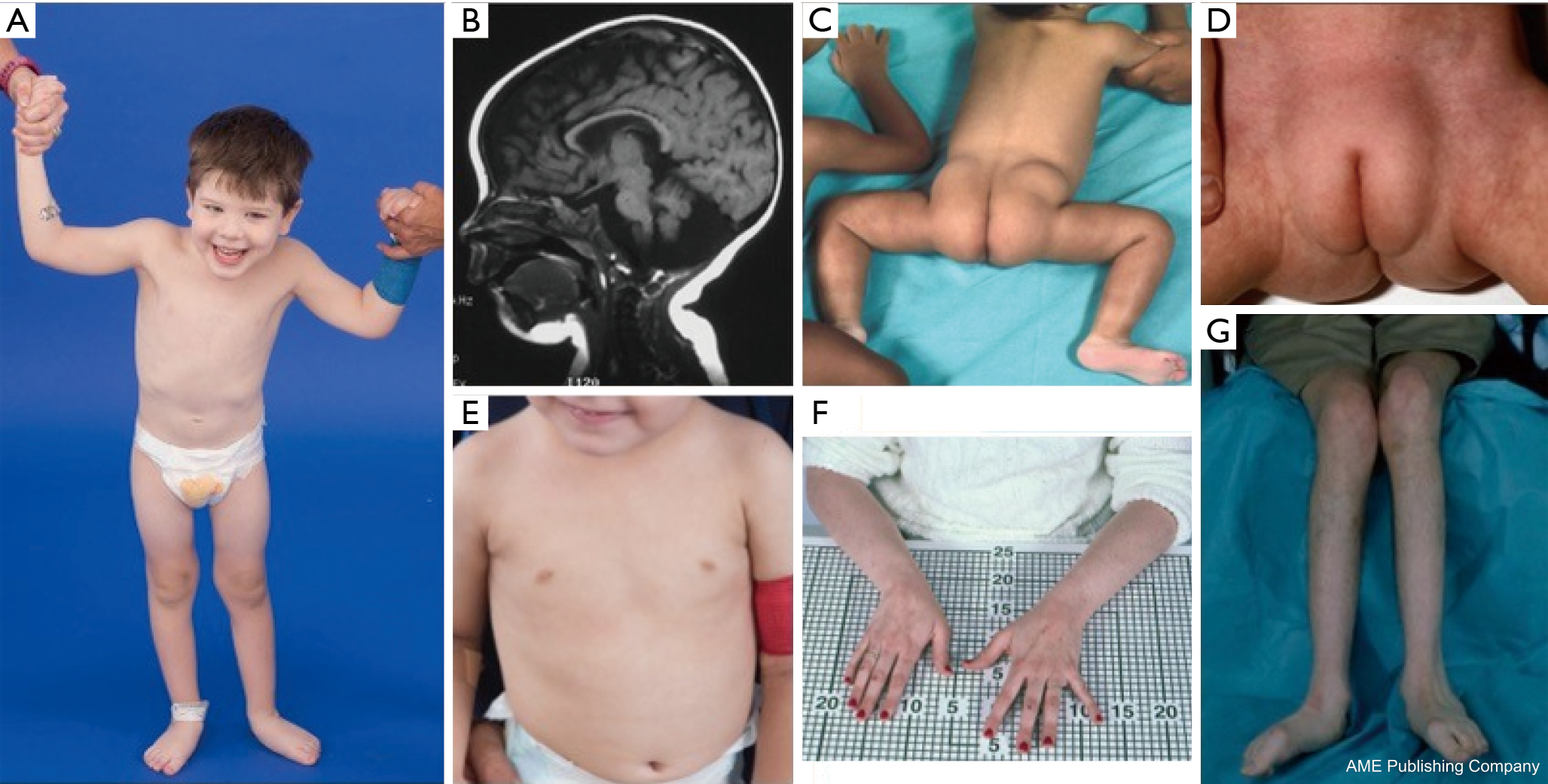
I barndommen kan de berørte personer udvikle retinitis pigmentosa, slagtilfælde-lignende episoder og kramper, tale- og motoriske forsinkelser og perifer neuropati. Konstitutionelt har patienterne almindeligvis manglende trivsel på grund af ernærings- og GI-anormaliteter og global udviklingsforsinkelse. Der kan observeres forhøjede levertransaminaser uden klinisk konsekvens, som typisk normaliseres ved 5 års alderen med lejlighedsvise udsving i forbindelse med sygdom (21,24). Leverbiopsier er sjældent indiceret ved CDG, medmindre der er mistanke om hepatisk fibrose (1). Klinisk hypothyroidisme er sjælden, men patienter med CDG bør få målt deres skjoldbruskkirtelhormoner og frit T4, hvilket kan vise lavt thyroidbindende globulin (TBG) og forbigående forhøjelser af thyroidstimulerende hormon (TSH) (37). Der er ikke rapporteret om misdannelser i lever og galdeveje hos PMM2-CDG-patienter.
Voksne med PMM2-CDG kan leve til deres 7. eller 8. årti med stabil kognitiv forsinkelse, perifer neuropati og progressiv thorakal og spinal kyfoskoliose med osteopeni eller osteoporose (34). Cerebellar ataksi er et stadig mere anerkendt symptom sammen med multisystemisk involvering (38-40). Endokrine abnormiteter, herunder hyperprolaktinæmi, frigivelse af væksthormon med hyperglykæmi, insulinresistens og hyperinsulinæmisk hypoglykæmi (41,42). Hos de berørte kvinder kan hypogonadotropisk hypogonadisme føre til manglende sekundær kønsudvikling eller manglende æggestokke (41,43,44). Patienterne kan have øget risiko for trombose på grund af nedsatte serumkoagulationsfaktorer, herunder faktor IV, IX og XI, antitrombin III, protein C og protein S (29).
MPI-CDG (CDG-Ib, mannosephosphat isomerase deficiency)
MPI-CDG er unik, fordi de berørte patienter har lille eller ingen neurologisk involvering, og nogle manifestationer af sygdommen kan behandles med oral mannose (2). Symptomerne er hovedsageligt hepatisk-intestinale uden dysmorfiske træk eller kognitive forsinkelser. Patienterne præsenterer sig typisk med tilbagevendende opkastninger, betydelig hypoglykæmi, manglende trivsel, potentielt livstruende proteinsvindingsenteropati, leverfibrotiske forandringer og dilatation af galdegangene (45-51). Patienterne har øget risiko for trombotiske hændelser på grund af lave serumkoncentrationer af protein C og S og antitrombin III.
ALG6-CDG (glucosyltransferase 1-mangel)
ALG6-CDG er den næsthyppigste N-glykosyleringsdefekt, der er karakteriseret ved lignende, men mildere fænotype end PMM2-CDG. Patienter med ALG6-CDG har manglende trivsel, udviklingsforsinkelse, hypotoni, kramper, strabismus, ataksi, koagulopati og dysmorfisme i ansigtet (dvs. lavtliggende ører, hypertelorisme og makroglossi). I lighed med MPI-CDG kan de også have proteinløsende enteropati. Desuden kan de berørte patienter have skeletanormaliteter, herunder brachydaktyli og fingermalformationer og skoliose. Affekterede patienter har typisk ikke typisk retinitis pigmentosa eller cerebellær hypoplasi (52).
O-linkede glykosyleringsdefekter og kombinerede N- og O-linkede glykosyleringsdefekter
På grund af den betydelige tilstedeværelse af O-glykaner i mucinholdige proteiner, herunder glykosaminoglykaner (GAG’er) og epiteloverflader (53), fører forstyrrelser i GAG-syntesen typisk til skeletdysplasier eller bindevævssygdomme. Påvirkede patienter kan præsentere muskuloskeletale, hud- og ledafvigelser (f.eks. ledslaksitet, multiple exostoser, chondro/osteosarkomer) ud over neurologiske symptomer (54-56). F.eks. O-glykosylaterer N-acetylgalactosaminyltransferase 3 (GALNT3) O-glykosylaterer det fosfaturskabende hormon FGF23, hvilket forhindrer proteolytisk spaltning og muliggør intakt sekretion. GALNT3-mangel fører til familiær tumorkalcinose, der er karakteriseret ved hyperfosfatæmi og ektopiske forkalkninger (57,58).
Defekter i glykosylering af lipider og GPI-ankerbiosyntese
Glykosfingolipider og deres sialylerede derivater, gangliosider, udtrykkes hovedsageligt af neuroner. Defekter i nedbrydningen af gangliosider fører til ophobning og de velkarakteriserede lysosomale lagersygdomme. I den modsatte ende er defekter i gangliosidbiosyntesen som f.eks. ST3GAL5-CDG og B4GALNT1-CDG yderst sjældne og fører til alvorlige neurodegenerative sygdomme. Patienterne kan vise sig med spastisk paraplegi, alvorlig intellektuel forsinkelse, epilepsi og ikke-neurologiske symptomer, herunder skeletdysplasi, dysmorfiske træk og unormal hudpigmentering (59,60).
Mutationer i mange gener inden for GPI-ankerbiosyntesevejen forårsager en række forskellige multiple medfødte anomalier, intellektuel funktionsnedsættelse og epilepsi. Den bedst karakteriserede GPI-biosyntesefejl, X-bunden PIGA-mangel, viser sig med spasmer hos spædbørn med hypsarytmi, hypotoni, flere hjerneabnormaliteter og ansigtsdysmorfismer. Patienterne kan også have varierende hud-, lever-, hjerte- og nyresygdomme (61-69). Nogle mutationer inden for PIGA forårsager den fænotypisk adskilte sygdom paroxysmal nocturnal hemoglobinuria (PNH), en erhvervet lidelse med knoglemarvssvigt (70,71).
Diagnose
Når der er klinisk mistanke om CDG, er det første skridt at bestille biokemisk CDG-testning i plasma eller serum, herunder CDT- og N-glykan-testning. Serum CDT- og N-glykananalyse kan kun påvise N-glykosyleringsdefekter, og de vil derfor ikke være nyttige til at differentiere isolerede O-glykosylerings- eller GPI-ankerdefekter. Transferrinisoformanalyse blev oprindeligt opnået ved isoelektrisk fokusering af transferrin, da manglende N-glykansyntese medfører delvis mangel på sialinsyre, hvilket ændrer ladningen på serumtransferrin og efterfølgende dets katodale migration på et elektroforisk felt. Massespektrometribaseret analyse af transferrin og N-glycan har imidlertid nu stort set erstattet isoelektrisk fokusering ved at identificere specifikke ændringer i oligosaccharider ved hjælp af masse og ladning (72).
N-bundne proteinglykosyleringsdefekter i proteiner
Serum transferrin CDT-resultater rapporteres som forholdet mellem mono-oligosakkarid/di-oligosakkarid transferrin, a-oligosaccharid/di-oligosaccharidtransferrin, tri-sialo/di-oligosaccharidtransferrin, apolipoprotein CIII-1/apolipoprotein CIII-2 og apolipoprotein CIII-0/apolipoprotein CIII-2-forholdet. Disse kvantitative resultater vil også komme med en fortolkning af fundmønstret.
Et type I-mønster transferrin CDT er karakteriseret ved øgede di- og asialotransferrinbånd og indikerer defekter i N-glykansyntesen i cytosol eller endoplasmatisk reticulum. Et type II-mønster er karakteriseret ved øgede di- og asialotransferrinbånd og tri- og/eller monosialotransferrinbånd og indikerer defekter i N-glykanprocessering i Golgi-apparatet (73).
Hvis der påvises et type I serum transferrin CDT-mønster, bør PMM2-mangel eller MPI-mangel være i forreste række ved differentiering, da PMM2-CDG er den mest almindelige CDG, og MPI-CDG kan behandles og er potentielt dødelig, hvis den ikke behandles. For at skelne mellem diagnoserne bør der foretages N-glykanprofilering, molekylær sekventering eller enzymatisk testning. Diagnosen PMM2-CDG eller MPI-CDG bekræftes ved molekylær testning, der viser bialleliske patogene varianter i PMM2 eller MPI, efterfulgt af PMM- eller MPI-enzymaktivitet i leukocytter eller fibroblaster, hvis patogeniciteten af genetiske varianter er usikker. N-glykananalyse eller molekylær analyse vil kunne differentiere størstedelen af ALG-CDG fra PMM2- eller MPI-CDG (15).
Et type II serum transferrin CDT-mønster indikerer Golgi-defekter såsom N-acetylglucosaminyltransferase (GnT) II-mangel (CDG type IIA, MGAT2-CDG). Apolipoprotein CIII (Apo-CIII)-isoformanalyse er en supplerende test for en type II CDT-profil, da den måler mucintype O-glykosyleringsdefekter i Golgi-apparatet. Der er begrænset følsomhed for CDT eller Apo-CIII med hensyn til påvisning af type II CDG. Der bør derfor foretages N-glykan- og O-glykanprofilering og molekylær panel- eller exomsekventering, når disse kliniske test er tilgængelige. Transferrin-glykosyleringsmønstre kan normaliseres sporadisk; gentagne test kan derfor være indiceret hos patienter med et højt mistankeindeks. Der kan opnås falske positive resultater hos patienter med akut krise af arvelig fructoseintolerance, galactosæmi, akut leversygdom og visse bakterielle infektioner. Ingen af de biokemiske CDG-tests kan screene for alle CDG’er, så selv i tilfælde af normale screeningsresultater kan der ved stærk klinisk mistanke foretages molekylær genpanelundersøgelse eller exomsekventering. Omvendt er biokemisk og funktionel bekræftelse af molekylærgenetiske fund også afgørende, da størstedelen af patienterne med CDG bærer mindst én mild og ofte ny missense-mutation.
O-linkede glykosyleringsdefekter og kombinerede N- og O-linkede glykosyleringsdefekter
Diagnosen er afhængig af molekylær sekventering, da transferrinisoformanalyse ikke ville opdage isolerede O-glykosyleringsdefekter. Kombinerede N- og O-linkede glykosyleringsdefekter kan påvises ved CDT, ApoCIII-analyse og plasma N-glykan- og O-glykananalyse.
Lipidglykosylerings- og GPI-ankerbiosyntesedefekter
Flowcytometri af blodgranulocytter måler celleoverfladeekspression af GPI-ankerede proteiner såsom CD16 og CD24. Flowcytometrianalyse af hvide blodlegemer eller røde blodlegemer for visse GPI-forankrede celleoverfladeproteiner er klinisk tilgængelig som en test for PNH som følge af erhvervede mutationer i PIGA-genet. PNH-testen kan afsløre abnormiteter i andre GPI-ankermangler, men diagnosen er for det meste afhængig af molekylær analyse.
Molekylær analyse
Det højeste diagnostiske udbytte for CDG er næste generations baseret gen-sekventeringspanel eller klinisk exom-sekventering (CES). Ved genetisk sekventering kontrolleres nucleotidsekvensen, eller “bogstav” stavningen, af gener for at afgøre, om der er en ændring, der påvirker genets funktion. Det menneskelige genom består af 3 millioner nukleotider, men kun 1-2 % af disse, kaldet exoner, bliver oversat til et funktionelt proteinprodukt. Det resterende ikke-kodende DNA, der er spredt mellem exonerne, og som ikke bliver oversat, kaldes introner (74). CES undersøger næsten alle kendte exoner af de ca. 20 000 gener i det menneskelige genom, som udgør et mindretal af det genetiske materiale i kromosomerne, men som højst sandsynligt indeholder sygdomsfremkaldende (patogene) varianter. CES kan også omfatte sekventering af mitokondrie-DNA (mtDNA), som afhører lille, ekstranukleært, cirkulært DNA, der befinder sig i mitokondrier, og som udelukkende er moderligt nedarvet.
De mulige resultater for CES omfatter positive, negative og varianter af ukendt betydning. Et positivt resultat betyder, at kendte sygdomsfremkaldende (dvs. patogene) varianter er identificeret, hvorefter diagnose, naturlig historie, prognose, risiko for tilbagefald og behandlingsmuligheder kan diskuteres. Et negativt resultat betyder, at der ikke blev identificeret nogen patogene varianter, der kan påvises. Varianter af ukendt betydning (også kaldet VUS) betyder, at selv om der blev identificeret genetiske ændringer, er der ikke tilstrækkelige oplysninger om den specifikke genetiske ændring til at vide med sikkerhed, om den er sygdomsfremkaldende. Der forventes variationer i hvert enkelt individs DNA, og derfor kan det hjælpe laboratoriet og den kliniske fortolkning af resultaterne at få forældreprøver testet samtidig med henblik på sammenligning. Det diagnostiske udbytte af CES skønnes at være 30-35 % og stiger med tiden, efterhånden som genopdagelsen og kendskabet til det menneskelige genom fortsætter med at udvikle sig (75-77). CES bestilles i stigende grad som det foretrukne førstevalg af bred genetisk testning på grund af den hurtige ekspeditionstid og de lave relative omkostninger i forhold til mængden af analyserede genetiske oplysninger. Begrænsningerne ved CES omfatter den manglende 100 % følsomhed, den manglende evne til at påvise visse typer genetiske ændringer (f.eks. deletioner, duplikationer, trinukleotidrepeats, dybe introniske mutationer eller methyleringsdefekter) og det forhold, at en diagnose muligvis ikke giver yderligere oplysninger om sygdommen eller ændrer håndteringen.
I rapporteringen af CES kan tilfældigt påviste patogene varianter i gener, der er forbundet med velkendte genetiske tilstande, rapporteres som sekundære fund (78). Denne liste over anbefalede sygdomme er kurateret af American College of Medical Genetics (ACMG). Genetic Information Nondiscrimination Act (GINA) er en vigtig overvejelse, når det skal besluttes, om man skal vælge at ind- eller udgå af at få kendskab til tilfældige fund (79). GINA beskytter enkeltpersoner mod misbrug af genetiske oplysninger i forbindelse med sygeforsikring og beskæftigelse, men ikke livsforsikring. GINA beskytter følgende genetiske oplysninger: familiens sygehistorie, bærerundersøgelser, prænatale genetiske undersøgelser, modtagelighed og prædiktive undersøgelser samt analyser af tumorer eller andre vurderinger af gen-, mutations- eller kromosomale ændringer.
Behandling
Behandlingen af CDG afhænger i høj grad af den enkeltes specifikke symptomer. Tilbagevendende symptomer hos patienter med CDG omfatter manglende trivsel, global udviklingsforsinkelse, opkastninger, slagtilfælde-lignende episoder og skeletanomalier. Klinisk eller subklinisk koagulopati, endokrinopati, hepatopati og kardiale defekter ses også ofte. Baseline-laboratorieundersøgelser for at fastslå sygdommens omfang og rutinemæssig overvågning anbefales, især for PMM2-CDG. Disse omfatter leverfunktionstest, serumalbumin, skjoldbruskkirtelfunktionstest, herunder frit T4, protein C, protein S, antithrombin III, faktor IX, urinanalyse og serumgonadotropiner og væksthormon.
Anbefalet billeddannelse omfatter ekkokardiogram, nyreultralyd, knoglealder, oftalmologisk undersøgelse med henblik på evaluering af linse, nethinde, okulær mobilitet og intraokulært tryk. Medmindre andet er angivet, anbefales rutinevaccinationer til voksne og børn, der er ramt af CDG. Antistoftitrer bør opgøres efter vaccination, da patienterne kan have et suboptimalt immunogent respons. Profylaktisk genopfyldning af koagulationsfaktorer før kirurgiske indgreb kan være nødvendig, hvis der er mangel ved baseline.
Der bør foretages en klinisk genetisk evaluering for at drøfte de arvelige aspekter af CDG samt etablere et medicinsk hjem for disse komplekse patienter. Det medicinske hjemsted er typisk den biokemiske genetiske afdeling, selv om genetik-, neuroudviklings- eller neurologiske afdelinger også har fungeret i denne egenskab, hvis der ikke er en dedikeret biokemisk genetisk afdeling til rådighed. Det er ofte nødvendigt at henvise til gastroenterologi, hæmatologi, endokrinologi, ernæringsstøtte, tale-, beskæftigelses-, fysio- og ernæringsterapi, ortopædkirurgi og rehabiliteringsmedicin.
Targeted terapier og prognose
Behandlingen af de fleste CDG-typer er stort set understøttende, med få undtagelser. MPI-CDG er den mest effektivt behandlelige af alle CDG. Oral mannose omdannes til mannose-6-fosfat af intracellulære hexokinaser, hvorved den enzymatiske blokering omgås og det mangelfulde substrat produceres. Mannosetilskud starter typisk med 1 g/kg kropsvægt pr. dag, fordelt på 4-6 doser pr. dag. Mens den potentielt livstruende proteinsvindingsenteropati er særlig lydhør over for mannosebehandling, kan leversygdommen i MPI-CDG fortsætte med at udvikle sig. De kliniske symptomer forbedres hurtigt, og transferrin CDT normaliseres i løbet af måneder, selv om leversygdommen kan fortsætte med at udvikle sig under behandlingen (45,80,81).
Forsigtighed bør udvises ved tilskud af mannose under graviditet, da indgift af mannose i drægtige hypomorfe fosfomannoseisomerase-musemodeller resulterede i embryonal letalitet og blindhed hos deres hvalpe (82). Desuden har intravenøs mannose været forbundet med nedsat bevidsthed og kramper, som forsvandt med glukoseadministration (83).
Behandlingen af PMM2-CDG er i vid udstrækning understøttende og baseret på symptomatologi. Imidlertid er kommende kliniske forsøg på mannose-1-fosfat-substraterstatningsterapi i øjeblikket under udvikling.
For anden CDG er forskellige orale simple sukkerarter blevet undersøgt med det formål teoretisk at forbedre hypoglykosyleringen. Fucose er blevet forsøgt for SLC35C1-CDG og galactose for PGM1-CDG og SLC35A2-CDG med blandede resultater (84). D-galactose på 1,0-2,5 g/kg/dag (maks. 50 gram) er blevet vist at forbedre hypoglykæmi, koagulopati og endokrinopati hos PGM1-CDG (85,86). Galaktose har også vist sig at forbedre endokrinopatien og koagulopatien hos TMEM165-CDG (87) og SLC39A8-CDG. Der blev også rapporteret om en betydelig klinisk forbedring hos SLC39A8-CDG-patienter på 15-20 mg/kg/dag MnSO4 (88). Der er kliniske forsøg i gang for at undersøge nytten af N-acetylmannosamin (ManNAc) i GNE-CDG (89), og flere prækliniske forsøg er i gang for andre CDG (90).
Trods medicinske fremskridt er der en betydelig dødelighed for børn med CDG inden for det første leveår som følge af multiorgansvigt eller alvorlig infektion (91). Spædbørn med CDG kan præsentere sig med fulminant multiorgansygdom, uhåndterbare kramper eller alvorlig hypoalbuminæmi, der udvikler sig til anasarki. Nogle patienter reagerer på aggressiv diurese og albuminerstatning, mens andre er refraktære over for behandling. Natriumbutyrat har vist sig at forbedre anfaldskontrollen hos CAD-CDG og PIGM-CDG (92). Ketogen diæt har også vist sig at mindske anfaldsfrekvensen i nogle tilfælde af PIGA-CDG (93). Under slagtilfælde-lignende episoder kan intravenøs hydrering og opretholdelse af normalt blodglukose være nyttigt, mens underliggende vaskulær trombotisk eller blødningsætiologi udelukkes.
Med fremkomsten af genomredigeringsteknikker og en bedre forståelse af mekanismen for sygdomme, der er omfattet af den diagnostiske paraply af CDG, er fremtiden for målrettet terapeutisk udvikling fortsat lovende.
Anerkendelser
Vi vil gerne takke Lynne Wolfe, ARNP og Donna Krasnewich, MD, PhD for at stille kliniske fotos til rådighed for os, som er opnået som en del af Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). Vi vil også gerne takke Jenny Thies, MS, LGC for hendes ekspertise inden for genetisk rådgivning.
Funding: IJ Chang er støttet af National Institutes of Health T32GM007454.
Fodnote
Interessekonflikter: Forfatterne har ingen interessekonflikter at oplyse.
Informeret samtykke: Forfatterne har ingen interessekonflikter at oplyse:
- Jaeken J, Matthijs G. Medfødte glykosyleringsforstyrrelser: Der blev indhentet skriftligt informeret samtykke fra patienterne til offentliggørelse af dette manuskript og eventuelle ledsagende billeder. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Medfødte glykosyleringsforstyrrelser: en hurtigt voksende sygdomsfamilie. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutationer i PMM2, der forårsager medfødte glykosyleringsforstyrrelser, type Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. Prævalensen af PMM2-CDG i Estland baseret på populationsbærende frekvenser og diagnosticerede patienter. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Manglende Hardy-Weinberg-ligevægt for den mest udbredte PMM2-mutation i CDG-Ia (congenital disorders of glycosylation type Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialinsyre-deficient serum og cerebrospinalvæske transferrin i et nyligt anerkendt genetisk syndrom. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG-nomenklatur: tid til en ændring! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Løsning af glykosyleringsforstyrrelser: grundlæggende tilgange afslører komplicerede veje. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Hvad er nyt inden for CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Medfødte glykosyleringsforstyrrelser. Del I. Defekter i protein N-glykosylering. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellulære funktioner af N-linkede glykaner. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Kliniske, laboratorie- og molekylære fund og langtidsopfølgningsdata hos 96 franske patienter med PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) samt gennemgang af litteraturen. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. En landsdækkende undersøgelse af PMM2-CDG i Italien: høj frekvens af en mild neurologisk variant, der er forbundet med L32R-mutationen. J Neurol 2015;262;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG i Sydafrika: Genotype-fænotypebeskrivelse af fem nye patienter. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Forskellige glykosyltransferaser er differentielt processeret til sekretion, dimerisering og autoglykosylering. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation (Medfødte glykosyleringsforstyrrelser). In: Mora-Montes H. Editor. Glycaner: Biochemistry, Characterization and Applications: Congenital Disorders of Glycosylation. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22;22:444-4.
- Rymen D, Jaeken J. Hudmanifestationer i CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF-system hos børn med medfødte glykosyleringsforstyrrelser. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. Et bredt spektrum af kliniske præsentationer ved medfødte glykosyleringsforstyrrelser I: en serie på 26 tilfælde. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Præpubertær vækst ved medfødt glykosyleringsforstyrrelse af type Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Kliniske og biokemiske karakteristika ved medfødt glykosyleringsforstyrrelse af type Ic, den første anerkendte endoplasmatisk retikulumdefekt i N-glykansyntese. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Medfødte glykosyleringsforstyrrelser: gennemgang af deres molekylære grundlag, kliniske præsentationer og specifikke terapier. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–en nyligt opdaget arvelig stofskiftesygdom. Multiple organmanifestationer, incidens 1/80.000, vanskelig at behandle. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Alvorlig forbigående myokardisk iskæmi forårsaget af hypertrofisk kardiomyopati hos en patient med en medfødt glykosyleringsforstyrrelse af type Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Kliniske træk hos voksne med medfødte glykosyleringsforstyrrelser af type Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Kulhydratdefekt glycoproteinsyndrom. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Kliniske og molekylære træk ved medfødt glykosyleringsforstyrrelse hos patienter med type 1 sialotransferrin-mønster og forskellig etnisk oprindelse. J Pediatr 2002;141;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc-modifikation af kappeproteinet fra potyvirus Plum pox virus forbedrer virusinfektion. Virology 2013;442;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Skandinaviske CDG-Ia-patienter: genotype/fænotypekorrelation og geografisk oprindelse af grundlæggermutationer. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 franske voksne patienter med PMM2-kongenital glykosyleringsforstyrrelse: resultat af den klassiske pædiatriske fænotype og beskrivelse af en sent indstillet fænotype. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinale karakteristika ved den medfødte medfødte glykosyleringsforstyrrelse PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Skjoldbruskkirtelfunktion i PMM2-CDG: diagnostisk tilgang og foreslået behandling. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental udvikling hos en patient med en medfødt glykosyleringsforstyrrelse (CDG) type Ia med multisystemisk involvering (intermediær fænotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Medfødt glykosyleringsforstyrrelse af type 1a: tre søskende med en mild neurologisk fænotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Nye forstyrrelser i kulhydratmetabolismen: medfødte glykosyleringsforstyrrelser og deres indvirkning på det endokrine system. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Medfødt glykosyleringsforstyrrelse af type Ia: heterogenitet i den kliniske præsentation fra multivisceral svigt til hyperinsulinæmisk hypoglykæmi som ledende symptomer hos tre spædbørn med fosfomannomutasemangel. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endokrinologi ved det kulhydratdeficiente glykoproteinsyndrom type 1 fra fødsel til ungdomsårene. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadal funktion og glykoproteinhormoner i kulhydratdefekt glykoproteinsyndromet (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Det kliniske spektrum af fosfomannoseisomerasemangel med en evaluering af mannosebehandling for CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrat-deficient glycoprotein syndrome type Ib. Phosphomannoseisomerasemangel og mannosebehandling. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Alvorlig hypoglykæmi som et symptom på carbohydratdeficient glycoproteinsyndrom. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinæmisk hypoglykæmi som et tegn på fosfomannoseisomerasemangel: En ny manifestation af kulhydratdefekt glycoproteinsyndrom, der kan behandles med mannose. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG6 defekt medfødt medfødt glykosyleringsforstyrrelse. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: Fra produktion til sekretion. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparansulfat i skeletudvikling, vækst og patologi: tilfælde af arvelige multiple exostoser. Dev Dyn 2013;242;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Udvidelse af det kliniske spektrum af B4GALT7-mangel: homozygot p.R270C-mutation med founder-effekt forårsager Larsen of Reunion Island-syndromet. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Forskellige ikke-inflammatoriske muskuloskeletale tilstande. Hyperfosfatematisk familiær tumoral calcinose (FGF23, GALNT3 og αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Ablation af Galnt3-genet fører til lave cirkulerende intakte koncentrationer af fibroblastvækstfaktor 23 (Fgf23) og hyperfosfatæmi på trods af øget Fgf23-ekspression. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. En mutation i et gangliosidbiosyntetisk enzym, ST3GAL5, resulterer i salt & peber syndrom, en neurokutan lidelse med ændret glykolipid- og glykoproteinglykosylering. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutationer i B4GALNT1 (GM2-syntase) ligger til grund for en ny lidelse i gangliosidbiosyntesen. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA-mutationer forårsager tidligt indsættende epileptiske encephalopatier og karakteristiske træk. Neurologi. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome er forårsaget af en mutation i PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorphisk promotormutation i PIGM forårsager arvelig glykosylphosphatidylinositolmangel. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorfe mutationer i PGAP2, der koder for et GPI-anker-remodellerende protein, forårsager autosomal-recessivt intellektuelt handicap. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. Fænotypen af en germlinemutation i PIGA: det somatisk muterede gen i paroxysmal natlig hæmoglobinuri. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutationer i PIGO, et medlem af GPI-ankersyntesevejen, forårsager hyperphosphatasi med mental retardering. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutationer i glycosylphosphatidylinositol-genet PIGL forårsager CHIME-syndromet. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. Det genotypiske og fænotypiske spektrum af PIGA-mangel. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) er forårsaget af somatiske mutationer i PIG-A-genet. EMBO J 1994;13:110-7.
- Brodsky RA. Paroxysmal natlig hæmoglobinuri. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. Massespektrometriens betydning for diagnosticering af medfødte glykosyleringsforstyrrelser. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessiv dilateret kardiomyopati på grund af DOLK-mutationer skyldes unormal O-mannosylering af dystroglycan O-mannosylering. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Fordeling af exoner og introner i det menneskelige genom. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Forbedret diagnostisk udbytte sammenlignet med målrettede gensekventeringspaneler tyder på en rolle for helgenomsekventering som en genetisk test på første niveau. Genet Med 2018;20;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. Omkostningerne og det diagnostiske udbytte af exomsekventering for børn med mistænkt genetiske lidelser: en benchmarkingundersøgelse. Genet Med 2018;20;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Anbefalinger for rapportering af sekundære fund i klinisk exom- og genomsekventering, 2016 opdatering (ACMG SF v2.0): en politikerklæring fra American College of Medical Genetics and Genomics. Genet Med 2017;19;19:249-55.
- Rothstein MA. Strømninger i moderne etik. GINA, ADA og genetisk forskelsbehandling i forbindelse med beskæftigelse. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Recidiverende tromboembolisme hos et barn med en medfødt glykosyleringsforstyrrelse (CDG) type Ib og behandling med mannose. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Udvikling af leversygdom på trods af mannosebehandling hos to patienter med CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannosetilskud inducerer embryonal letalitet og blindhed i fosfomannoseisomerase-hypomorfe mus. FASEB J 2014;28;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Krampeanfald og stupor under intravenøs mannosebehandling hos en patient med CDG-syndrom type 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Fucosetilskud ved leukocytadhæsionsmangel type II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Arvelig glycosylphosphatidylinositolmangel: en CDG, der kan behandles. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Oralt tilskud af D-galaktose i PGM1-CDG. Genet Med 2017;19;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Galaktosetilskud hos patienter med TMEM165-CDG redder glykosyleringsdefekter. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8-mangel: biokemisk korrektion og stor klinisk forbedring ved manganbehandling. Genet Med 2018;20;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Orale monosakkaridterapier til at vende renal og muskelhyposialylering i en musemodel af GNE-myopati i en musemodel af GNE-myopati. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: Fra bænk til sengekant. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Menneskelige glykosyleringsforstyrrelser. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Målrettet terapi for arvelig GPI-mangel. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogen diæt – en ny behandling af tidlig epileptisk encephalopati som følge af PIGA-mangel. Brain Dev 2016;38;38:848-51.