- Overview
- Epidemiologie
- Clasificare biochimică și nomenclatură
- Genetică
- Pathophysiology
- Defectele de glicozilare a proteinelor legate de N
- Defecte de glicozilare legată de O și defecte combinate de glicozilare legată de N și O
- Defecte de glicozilare a lipidelor și de biosinteză a ancorelor GPI
- Manifestări clinice
- Defecte de glicozilare a proteinelor N-legate
- PMM2-CDG (CDG-Ia, deficit de PMM2)
- MPI-CDG (CDG-Ib, deficit de mannofosfat izomerază)
- ALG6-CDG (deficit de glucoziltransferază 1)
- Defecte de glicozilare legată de O și defecte combinate de glicozilare legată de N și O
- Defecte de biosinteză a glicozilării lipidelor și a ancorelor GPI
- Diagnostic
- Defecte de glicozilare a proteinelor N-legate
- Defecte de glicozilare legate de O și defecte combinate de glicozilare legate de N și O
- Defecte de glicozilare a lipidelor și de biosinteză a ancorelor GPI
- Analiză moleculară
- Management
- Terapii țintite și prognostic
- Recunoștințe
- Nota de subsol
Overview
Glicozilarea este procesul de adăugare a reziduurilor de zahăr la proteine și lipide în diferite căi celulare. Tulburările congenitale de glicozilare (CDG) reprezintă un grup eterogen din punct de vedere genetic și clinic de peste o sută de boli cauzate de defecte în diferite etape de-a lungul căilor de sinteză sau de modificare a glicanilor. Cele mai multe dintre aceste boli monogenice au o moștenire autosomal recesivă, dar au fost descrise și forme autosomal dominante și legate de X.
DCDG se prezintă de obicei cu manifestări multisistemice, cel mai frecvent cu întârzieri în dezvoltare, incapacitate de creștere, hipotonie, anomalii neurologice, hepatopatie și coagulopatie. Persoanele afectate pot prezenta, de asemenea, afecțiuni oculare, cutanate și cardiace, precum și dismorfisme faciale. Deși modificările neurologice și întârzierile cognitive sunt observate la majoritatea indivizilor afectați, există anumite cazuri și chiar tipuri care nu au manifestări neurologice. Având în vedere etiologia clinică și genetică largă a CDG, diagnosticul clinic se bazează pe un indice ridicat de suspiciune în boala multisistemică.
Analiza transferinei cu deficit de carbohidrați în ser (CDT) este testul de screening de primă linie la pacienții cu suspiciune de CDG, dar este limitată în detectarea defectelor de N-glicozilare cu deficiențe de acid sialic. Testele de linia următoare includ analiza glicanilor legați de dolicoli și testele genetice. Diagnosticul precoce al acestui grup de boli în creștere exponențială este important, deoarece unele CDG sunt tratabile. Tratamentul pentru defectele de glicozilare este în principal de susținere, deși sunt disponibile terapii țintite pentru MPI-CDG, SLC35C1-CDG, PIGM-CDG și PGM1-CDG. Detalii despre aceste tratamente se găsesc în secțiunea „Terapii țintite și prognostic” de mai jos. Accentul acestei revizuiri se va pune pe cele mai frecvente tipuri de CDG cu fenotipuri sau tratamente recognoscibile, publicul țintă fiind furnizorii de asistență medicală primară.
Epidemiologie
Incidența și prevalența tuturor tipurilor de CDG în ansamblu nu au fost bine stabilite, deși au fost raportați pacienți la nivel mondial din aproape toate mediile etnice și ambele sexe sunt afectate în mod egal. Prevalența estimată în populațiile europene și afro-americane este de 1/10.000 pe baza frecvențelor de purtători ai variantelor patogene cunoscute în 53 de gene (1-4). Prevalența celui mai frecvent diagnosticat CDG, PMM2-CDG, variază de la 1/20.000 în populațiile olandeze și 1/77.000 în Estonia, pe baza unor rapoarte izolate (5,6). Până în prezent, au fost raportate mai puțin de 100 de cazuri pentru majoritatea tipurilor de CDG.
Clasificare biochimică și nomenclatură
În general, CDG sunt clasificate în prezent în patru categorii – (I) glicozilare legată de N, (II) glicozilare legată de O, (III) glicozilare combinată legată de N și O/glicozilare multiplă și (IV) defecte de biosinteză a ancorelor de lipide și glicozilfosfatidilinositol (GPI).
Defectul de glicozilare a proteinelor N-legate PMM2-CDG, cunoscut anterior sub numele de CDG tip Ia, a fost primul CDG raportat de Jaeken în 1980 și rămâne de departe cel mai frecvent CDG până în prezent (7). PMM2-CDG a fost denumită inițial „sindromul glicoproteinei cu deficit de carbohidrați” din cauza multiplelor anomalii ale glicoproteinei serice observate prin focalizarea izoelectrică a transferinei serice la persoanele afectate. Din punct de vedere istoric, CDG au fost clasificate în funcție de modelele de analiză a izoformelor de transferrină – modelele de tip I au fost atribuite defectelor de asamblare și transfer ale glicanilor legați de dolicoli, localizate în citoplasmă sau în ER, iar modelele de tip II au fost atribuite defectelor de procesare în aparatul Golgi. De la acest punct de ramificare, CDG-urile au fost apoi denumite în ordine alfabetică, în ordinea descoperirii.
Cu apariția diagnosticelor moleculare pe scară largă, nomenclatura CDG-urilor a fost actualizată în 2008 pentru a specifica etiologia moleculară a bolii, reflectând creșterea exponențială a căilor și tulburărilor care nu se încadrau perfect în categoriile dihotomice stabilite anterior. În prezent, nomenclatura CDG este indicată prin numele genei afectate (fără litere italice, nume de gene la www.genenames.org), urmat de -CDG (de exemplu, PMM2-CDG) (8).
Genetică
Marea majoritate a tulburărilor congenitale de glicozilare sunt moștenite în mod autosomal recesiv, cu o mutație moștenită de la fiecare părinte asimptomatic (purtător). Testarea moleculară, de obicei cu metode de secvențiere de generație următoare, este necesară pentru a stabili un diagnostic genetic. Testarea părinților pentru varianta cunoscută poate confirma moștenirea versus apariția de novo. Pentru moștenirea autozomal recesivă, riscul de recurență pentru frați și pentru fiecare sarcină a unui individ afectat este de 25% pentru a fi afectat, 50% pentru a fi purtător asimptomatic și 25% pentru a nu fi afectat.
O mână de CDG au moștenire autozomal dominantă (N-linked: GANAB-CDG, PRKCSH-CDG; O-linked: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Mai puține sunt legate de X (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). Majoritatea formelor dominante și unele forme de CDG legate de X se datorează unor mutații de novo. Bolile și genele specifice sunt descrise mai jos în secțiunea „fiziopatologie”.
Datele de mutație pentru toate genele publicate pentru CDG sunt disponibile la Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Informații despre variantele genetice specifice sunt disponibile la Leiden Open Variation Database cu instrumente integrate de patogenitate in silico (http://www.lovd.nl/3.0/home). Sinopsisuri clinice pentru gene specifice pot fi găsite la Online Mendelian Inheritance in Man (http://www.omim.org/) sau, într-un domeniu mai limitat, la GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Având în vedere numărul mic de pacienți afectați pentru majoritatea subtipurilor de CDG, corelația genotip-fenotip este dificil de stabilit.
Pathophysiology
Peste 130 de tipuri de CDG au fost raportate până în prezent (9,10). Având în vedere prezența omniprezentă a căilor de glicozilare, CDG sunt extrem de diverse în patogeneza lor biochimică. Numeroase proteine și lipide (de exemplu, sfingolipide și glicolipide) suferă glicozilare cu monosacaride și/sau oligozaharide, denumite colectiv glicani, în diferite compartimente celulare. Locațiile lor subcelulare sunt diverse, dar cele mai multe defecte apar în cadrul ER sau al aparatului Golgi. Caracteristicile clinice și etiologia genetică a celor mai frecvente CDG în funcție de cale sunt rezumate în tabelul S1.
Între proteine, glicanii sunt descriși prin legătura lor cu lanțul polipeptidic -N-glicanii sunt atașați de grupa amidă a asparaginei (Asn), în timp ce O-glicanii sunt atașați de grupa hidroxil fie a serinei, fie a treoninei. Sinteza N-glicanilor necesită construirea etapizată a zaharurilor legate de nucleotide în citosol, asamblarea în reticulul endoplasmatic și procesarea în aparatul Golgi. În schimb, sinteza O-glicanilor necesită asamblare, dar nu și procesare, prin urmare, defectele de O-glicozilare apar predominant în aparatul Golgi.
Defectele de glicozilare a proteinelor legate de N
N-glicozilarea implică atașarea covalentă a structurilor de carbohidrați la gruparea amidică a lanțului lateral al reziduurilor Asn în cadrul unui site acceptor consensual Asn-X-Ser/Thr, translocarea polipeptidei substrat în reticulul endoplasmatic pentru remodelare și modificarea ulterioară a lanțului N-glican în cadrul aparatului Golgi (11,12). Defectele în orice punct de-a lungul căii de sinteză, asamblare și procesare pot duce la boală clinică.
PMM2-CDG este cauzată de variante patogene în gena fosfomanomutazei 2 (PMM2), ceea ce duce la deficiența enzimei PMM2 care catalizează conversia citosolică a mannozei-6-fosfat în mannoză-1-fosfat în a doua etapă a sintezei mannozei de guanozină difosfat (GDP). Majoritatea pacienților găzduiesc mutații missense patogene heterozigote compuse (www.lovd.nl/PMM2). Cea mai frecventă variantă patogenă recurentă p.Arg141His se găsește la aproximativ 40 % dintre persoanele afectate de origine europeană, iar p.Phe119Leu este, de asemenea, frecvent întâlnită în nordul Europei (1). Au fost raportate corelații genotip-fenotip pentru PMM2-CDG (3,13,14).
MPI-CDG este o tulburare autozomal recesivă cauzată de variante patogene în gena mannoză fosfat izomerazei (MPI) care duce la o isomerază fosfomanozică (MPI) deficitară. MPI catalizează în mod normal prima etapă a sintezei GDP-mannoză (adică transformarea fructozei-6-fosfat în mannoză-6-fosfat), dar fructoza-6-fosfat nu se acumulează intracelular, deoarece poate fi metabolizată și prin calea glicolitică. Prin urmare, deși similar din punct de vedere biochimic cu PMM2-CDG, MPI-CDG nu provoacă o implicare neurologică și multisistemică la fel de semnificativă. CDT este, de asemenea, testul de screening de elecție pentru MPI-CDG, care prezintă un model de tip 1. Diagnosticul poate fi apoi confirmat pe cale moleculară sau prin activitatea MPI a fibroblastelor/leucocitelor.
ALG6-CDG este o boală recesivă cauzată de mutații în ALG6, ceea ce duce la atașarea anormală a trei molecule de glucoză la intermediari de mannoză legați de dolicoli și hipoglicozilarea în aval a glicoproteinelor serice (15).
Defecte de glicozilare legată de O și defecte combinate de glicozilare legată de N și O
O-glicozilarea cuprinde adăugarea în trepte a lanțurilor de carbohidrați la reziduurile de serină, treonină și hidroxilizină ale proteinelor de către glicoziltransferazele din aparatul Golgi (16). Mai multe tipuri de glicani legați O au fost asociate cu boli umane, denumite după primul zahăr atașat la reziduul de aminoacid (17).
Defecte de glicozilare a lipidelor și de biosinteză a ancorelor GPI
Ancorele GPI sunt glicolipide care suferă asamblare secvențială în reticulul endoplasmatic și modificări în cadrul aparatului Golgi. Tulburările de biosinteză a ancorelor GPI datorate deficiențelor enzimatice sunt denumite în ordine alfabetică după ordinea descoperirii și nu cronologic în funcție de etapa de asamblare. Odată sintetizate, ancorele GPI rezidă pe membranele plasmatice și se leagă de sute de proteine de pe suprafața celulară, îndeplinind o multitudine de funcții celulare. Cele mai multe dintre aceste boli sunt autosomal recesive, cu excepția notabilă a deficitului de PIGA legat de X.
Manifestări clinice
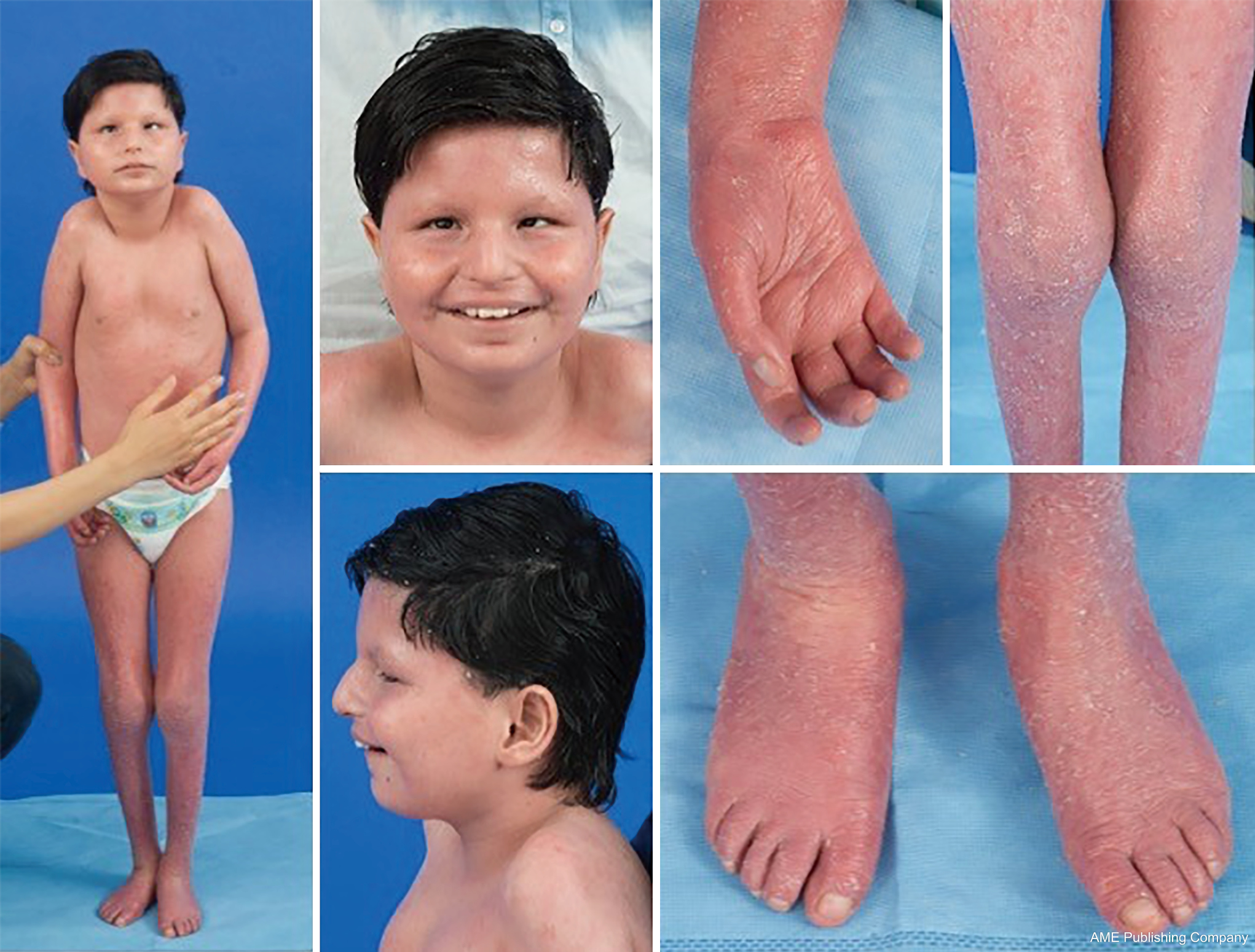
Datorită prezenței omniprezente a căilor de glicozilare, practic orice sistem de organe poate fi implicat în CDG, deși majoritatea cazurilor implică anomalii neurologice. Unele CDG se prezintă cu ihtioză, inclusiv MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (figura 1) și PIGL-CDG (18,19). Aproape toate CDG prezintă o boală multisistemică în primii ani de viață, cu excepția unora care afectează doar un singur sistem de organe (de ex, retina în DHDDS-CDG; joncțiunea neuromusculară în ALG2-CDG, ALG14-CDG, CFPT1-CDG; creierul în ST3GAL3-CDG, TUSC3-CDG; pielea sau mușchii scheletici în POGLUT1-CDG, POFUT1-CDG; cartilajul în EXT1/EXT2-CDG; ficatul în TMEM199-CDG; celulele roșii din sânge în SEC23B-CDG). Vârsta de apariție și severitatea pot varia de la letalitatea neonatală până la vârsta adultă aproape asimptomatică și orice permutare între acestea. Cea mai frecvent raportată constelație de simptome include întârzierea dezvoltării, incapacitatea de a se dezvolta, hipotonie, anomalii neurologice, hipoglicemie și anomalii hepatice, oculare, cutanate, gastrointestinale, imunologice, scheletice și de coagulare variabile (19).
Fenotipul complet pentru multe subtipuri CDG rămâne să fie pe deplin delimitat din cauza rarității cazurilor raportate. Prin urmare, CDG ar trebui să fie luată în considerare în orice context de boală multisistemică, în special în cazurile cu o componentă neurologică sau întârziere nespecifică a dezvoltării cu etiologie neclară.
Deși fiziopatologia simptomelor multitudinii rămâne să fie elucidată, a fost clarificată relația dintre anumite căi de glicozilare și simptome clinice specifice. De exemplu, incapacitatea de a se dezvolta observată în multe tipuri de CDG este atribuită hipoglicozilării și formării deficitare a mai multor glicoproteine din cadrul căii de creștere a insulinei, inclusiv IGF-1, ALS și IGFBP-3 (20). Pe măsură ce înțelegem mai bine acest grup de tulburări complexe, CDG sunt din ce în ce mai mult recunoscute la persoanele cu diagnostice evazive. Implicările sistemului de organe ale diferitelor CDG sunt rezumate în tabelul S1. Vom discuta mai jos caracteristicile clinice ale celor mai frecvente forme și forme cu tratamente țintite de CDG.
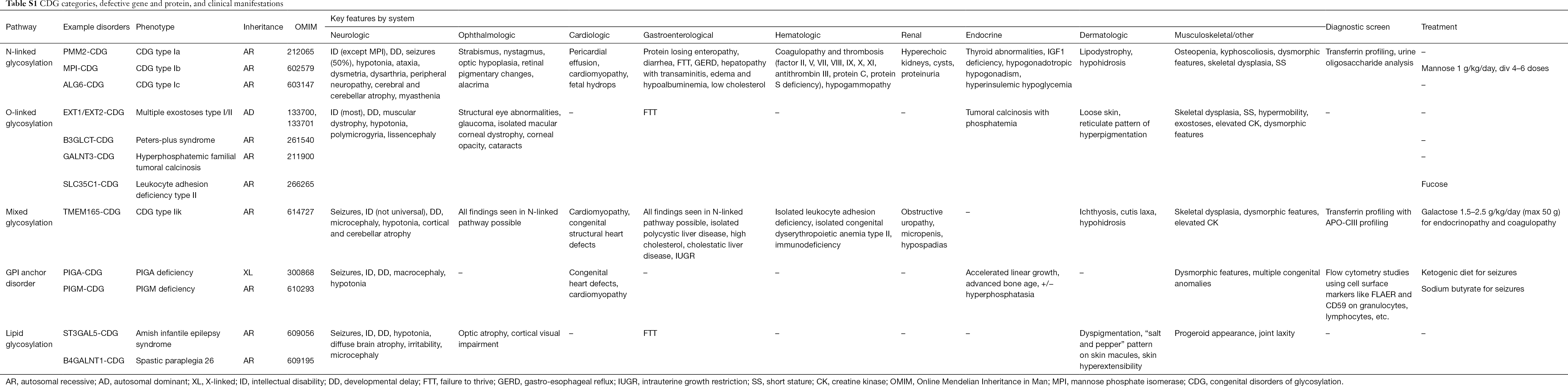
Tabel complet
Defecte de glicozilare a proteinelor N-legate
Ca cele mai frecvent diagnosticate CDG, fenotipul tulburărilor de glicozilare N-legate este adesea anunțat ca fiind prezentarea clasică. Cu toate acestea, spectrul fenotipic al CDG este destul de divers, iar multe CDG pot să nu se prezinte cu simptome stereotipice asociate cu PMM2-CDG.
PMM2-CDG (CDG-Ia, deficit de PMM2)
PMM2-CDG este cea mai frecventă CDG, cu peste 700 de cazuri raportate la nivel mondial. Se caracterizează prin boală multisistemică severă în copilărie, boală neurologică și întârziere în dezvoltare în copilărie și/sau dizabilitate intelectuală stabilă la vârsta adultă (21,22).
În copilărie, PMM2-CDG se prezintă cu anomalii neurologice de obicei la scurt timp după naștere, și anume strabism și mișcări oculare anormale, hipoplazie cerebeloasă, hipotonie, retard psihomotor, ataxie, hipotonie și hiporeflexie. Sugarii pot prezenta, de asemenea, afecțiuni hepatice, sindrom nefrotic și chisturi renale, revărsare pericardică și cardiomiopatie hipertrofică, insuficiență de dezvoltare și insuficiență multiorganică care duce la deces în primul an de viață la până la 20% dintre persoanele afectate (21,23-28).
O constelație de trăsături dismorfice a fost descrisă la pacienții cu PMM2-CDG (figurile 2,3). Acestea includ un cerebel hipoplastic, dismorfisme faciale (de exemplu, urechi mari, displazice), mameloane inversate și o distribuție anormală a țesutului adipos pe fese sau în regiunea suprapubiană, care se poate rezolva cu vârsta (14,21,29-32). Pacienții au fost descriși ca având un comportament sociabil și vesel. Prezentarea este foarte variabilă, deși strabismul poate fi observat la mai mult de 70% dintre pacienții afectați (21,23,33-35). Mameloanele inversate și tampoanele adipoase anormale sunt observate la aproximativ 25-50% dintre pacienți (36).

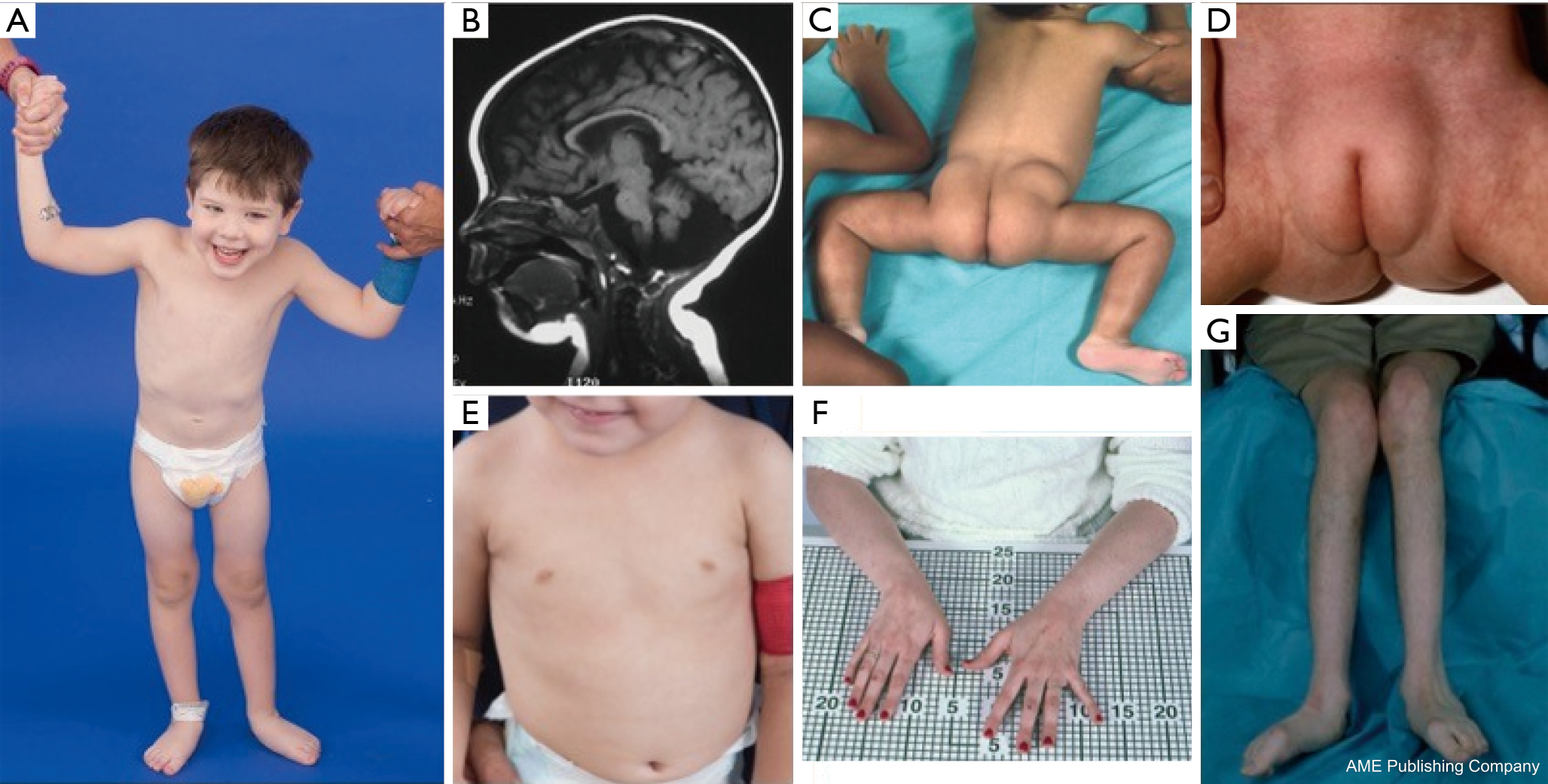
În copilărie, persoanele afectate pot dezvolta retinită pigmentară, episoade asemănătoare unui accident vascular cerebral și convulsii, întârzieri de vorbire și motorii și neuropatie periferică. Din punct de vedere constituțional, pacienții prezintă în mod obișnuit un eșec de creștere din cauza anomaliilor alimentare și gastrointestinale și o întârziere globală a dezvoltării. Se pot observa transaminaze hepatice crescute fără consecințe clinice, care se normalizează de obicei până la vârsta de 5 ani, cu fluctuații ocazionale în funcție de boală (21,24). Biopsiile hepatice sunt rareori indicate în CDG, cu excepția cazului în care se suspectează fibroza hepatică (1). Hipotiroidismul clinic este rar, dar pacienților cu CDG trebuie să li se măsoare hormonii tiroidieni și T4 liber, care pot arăta globulină de legare a tiroidei (TBG) scăzută și creșteri tranzitorii ale hormonului de stimulare a tiroidei (TSH) (37). Malformațiile hepatice și ale căilor biliare nu au fost raportate la pacienții cu PMM2-CDG.
Adulții cu PMM2-CDG pot trăi până în deceniile 7 sau 8 cu întârziere cognitivă stabilă, neuropatie periferică și cifoscolioză toracică și spinală progresivă cu osteopenie sau osteoporoză (34). Ataxia cerebeloasă este un simptom din ce în ce mai recunoscut, împreună cu implicarea multisistemică (38-40). Anomalii endocrine, inclusiv hiperprolactinemie, eliberarea hormonului de creștere cu hiperglicemie, rezistență la insulină și hipoglicemie hiperinsulinemică (41,42). La femeile afectate, hipogonadismul hipogonadotropic poate duce la absența dezvoltării sexuale secundare sau la absența ovarelor (41,43,44). Pacienții pot prezenta un risc crescut de tromboză din cauza scăderii factorilor serici de coagulare, inclusiv a factorilor IV, IX și XI, a antitrombinei III, a proteinei C și a proteinei S (29).
MPI-CDG (CDG-Ib, deficit de mannofosfat izomerază)
MPI-CDG este unică, deoarece pacienții afectați au o implicare neurologică mică sau deloc și unele manifestări ale bolii sunt tratabile prin mannoză orală (2). Simptomele sunt în principal hepatico-intestinale, fără caracteristici dismorfice sau întârzieri cognitive. Pacienții prezintă în mod obișnuit vărsături recurente, hipoglicemie semnificativă, incapacitate de creștere, enteropatie cu pierdere de proteine care poate pune viața în pericol, modificări fibrotice hepatice și dilatare a căilor biliare (45-51). Pacienții prezintă un risc crescut de apariție a evenimentelor trombotice din cauza concentrațiilor serice scăzute de proteină C și S și de antitrombină III.
ALG6-CDG (deficit de glucoziltransferază 1)
ALG6-CDG este al doilea cel mai frecvent defect de N-glicozilare caracterizat printr-un fenotip similar, dar mai blând decât PMM2-CDG. Pacienții cu ALG6-CDG au eșec de creștere, întârziere în dezvoltare, hipotonie, convulsii, strabism, ataxie, coagulopatie și dismorfisme faciale (de exemplu, urechi joase, hipertelorism și macroglosie). Similar cu MPI-CDG, aceștia pot avea, de asemenea, enteropatie cu pierdere de proteine. În plus, pacienții afectați pot avea anomalii ale scheletului, inclusiv brahidacizie și malformații ale degetelor și scolioză. Pacienții afectați nu au în mod obișnuit retinită pigmentară sau hipoplazie cerebeloasă (52).
Defecte de glicozilare legată de O și defecte combinate de glicozilare legată de N și O
Datorită prezenței substanțiale a O-glicanilor în proteinele care conțin mucină, inclusiv în glicozaminoglicani (GAG) și în suprafețele epiteliale (53), tulburările de sinteză a GAG duc de obicei la displazii scheletice sau la boli ale țesutului conjunctiv. Pacienții afectați pot prezenta anomalii musculo-scheletice, cutanate și articulare (de exemplu, laxitate articulară, exostoze multiple, condro/osteosarcoame) în plus față de simptome neurologice (54-56). De exemplu, N-acetilgalactosaminiltransferaza 3 (GALNT3) O-glicozilează hormonul fosfatural, FGF23, împiedicând scindarea proteolitică și permițând secreția sa intactă. Deficiența GALNT3 duce la calcinoză tumorală familială, caracterizată prin hiperfosfatemie și calcificări ectopice (57,58).
Defecte de biosinteză a glicozilării lipidelor și a ancorelor GPI
Glicozfolipidele și derivații lor sialiați, gangliozidele, sunt exprimate în principal de neuroni. Defectele în descompunerea gangliozidelor conduc la acumularea și la bolile de depozitare lizozomală bine caracterizate. La polul opus, defectele de biosinteză a gangliozidelor, cum ar fi ST3GAL5-CDG și B4GALNT1-CDG, sunt extrem de rare și conduc la boli neurodegenerative grave. Pacienții pot prezenta paraplegie spastică, întârziere intelectuală severă, epilepsie și simptome non-neurologice, inclusiv displazie scheletală, trăsături dismorfice și pigmentare anormală a pielii (59,60).
Mutațiile în multe gene din cadrul căii de biosinteză a ancorelor GPI cauzează o varietate de anomalii congenitale multiple, dizabilitate intelectuală și epilepsie. Cel mai bine caracterizat defect de biosinteză a GPI, deficitul de PIGA legat de X, se prezintă cu spasme infantile cu hipsaritmie, hipotonie, multiple anomalii cerebrale și dismorfisme faciale. Pacienții pot prezenta, de asemenea, afecțiuni cutanate, hepatice, cardiace și renale variabile (61-69). Unele mutații în cadrul PIGA cauzează boala distinctă din punct de vedere fenotipic hemoglobinurie paroxistică nocturnă (PNH), o tulburare dobândită de insuficiență medulară (70,71).
Diagnostic
Când se suspectează clinic o CDG, primul pas este să se solicite testarea biochimică a CDG în plasmă sau ser, inclusiv CDT și testarea N-glicanilor. Analiza CDT și a N-glicanilor din ser pot detecta doar defectele de N-glicozilare, prin urmare nu ar fi utile pentru a diferenția defectele izolate de O-glicozilare sau de ancorare GPI. Analiza izoformelor de transferrină a fost obținută inițial prin focalizarea izoelectrică a transferrinei, deoarece eșecul sintezei N-glicanilor determină o deficiență parțială a acidului sialic, care modifică sarcina transferrinei serice și, ulterior, migrarea sa catodică pe un câmp electroforetic. Cu toate acestea, analiza bazată pe spectrometria de masă a transferrinei și a N-glicanului a înlocuit în prezent în mare măsură focalizarea izoelectrică prin identificarea modificărilor specifice ale oligozaharidelor în funcție de masă și sarcină (72).
Defecte de glicozilare a proteinelor N-legate
Rezultatele CDT ale transferrinei serice sunt raportate ca raport mono-oligozaharide/di-oligozaharide ale transferrinei, a-oligozaharide/di-oligozaharide transferrină, tri-sialo/di-oligozaharide transferrină, raportul apolipoproteină CIII-1/apolipoproteină CIII-2 și raportul apolipoproteină CIII-0/apolipoproteină CIII-2. Aceste rezultate cantitative vor veni, de asemenea, cu o interpretare a tiparului constatărilor.
Un tipar de tip I al CDT de transferrină se caracterizează prin creșterea benzilor de di- și asialotransferrină și indică defecte în sinteza de N-glicani în citosol sau în reticulul endoplasmatic. Un tipar de tip II se caracterizează prin benzi crescute de di- și asialotransferină și benzi de tri- și/sau monosialotransferină și indică defecte în procesarea N-glicanilor în aparatul Golgi (73).
Dacă se detectează un tipar CDT de transferrină serică de tip I, deficitul de PMM2 sau deficitul de MPI ar trebui să fie în fruntea diferențialelor, deoarece PMM2-CDG este cea mai frecventă CDG, iar MPI-CDG este tratabilă și potențial fatală dacă este lăsată netratată. Pentru a diferenția între diagnostice, ar trebui să se efectueze profilarea N-glicanilor, secvențierea moleculară sau teste enzimatice. Diagnosticul de PMM2-CDG sau MPI-CDG este confirmat prin teste moleculare care arată variante patogene bialelice în PMM2 sau MPI, urmate de activitatea enzimatică PMM sau MPI în leucocite sau fibroblaste dacă patogenitatea variantelor genetice este incertă. Analiza N-glicanilor sau analiza moleculară ar diferenția majoritatea ALG-CDG de PMM2 sau MPI-CDG (15).
Un model CDT de transferină serică de tip II indică defecte Golgi, cum ar fi deficitul de N-acetilglucozaminiltransferază (GnT) II (CDG tip IIA, MGAT2-CDG). Analiza izoformei apolipoproteinei CIII (Apo-CIII) este un test complementar pentru un profil CDT de tip II, deoarece măsoară defectele de glicozilare O de tip mucină în aparatul Golgi. Există o sensibilitate limitată pentru CDT sau Apo-CIII în detectarea CDG de tip II. Astfel, profilarea N-glicanilor și O-glicanilor și panoul molecular sau secvențierea exomului ar trebui să fie întreprinse atunci când aceste teste clinice sunt disponibile. Tiparele de glicozilare a transferinei se pot normaliza sporadic; prin urmare, testele repetate pot fi indicate la pacienții cu un indice ridicat de suspiciune. Pot fi obținute rezultate fals pozitive la pacienții cu crize acute de intoleranță ereditară la fructoză, galactosemie, boală hepatică acută și unele infecții bacteriene. Niciunul dintre testele biochimice pentru CDG nu poate depista toate CDG-urile, astfel încât, chiar și în prezența unor rezultate de screening normale, se poate efectua testarea panelului genetic molecular sau secvențierea exomului în cazul unei suspiciuni clinice puternice. În schimb, confirmarea biochimică și funcțională a rezultatelor genetice moleculare este, de asemenea, esențială, deoarece majoritatea pacienților cu CDG sunt purtători a cel puțin unei mutații missense ușoare și adesea noi.
Defecte de glicozilare legate de O și defecte combinate de glicozilare legate de N și O
Diagnosticul se bazează pe secvențierea moleculară, deoarece analiza izoformei transferinei nu ar detecta defectele izolate de glicozilare O. Defectele combinate de glicozilare legate de N și O pot fi detectate prin CDT, analiza ApoCIII și analiza glicanilor N și O din plasmă.
Defecte de glicozilare a lipidelor și de biosinteză a ancorelor GPI
Citometria în flux a granulocitelor sanguine măsoară expresia suprafeței celulare a proteinelor ancorate de GPI, cum ar fi CD16 și CD24. Analiza prin citometrie în flux a globulelor albe sau a globulelor roșii pentru anumite proteine de suprafață celulară ancorate de GPI este disponibilă clinic ca test pentru PNH datorată mutațiilor dobândite în gena PIGA. Testul pentru PNH poate evidenția anomalii în alte deficiențe ale ancorelor GPI, dar diagnosticul se bazează în principal pe analiza moleculară.
Analiză moleculară
Cel mai mare randament de diagnostic pentru CDG este panoul de secvențiere genetică bazat pe următoarea generație sau secvențierea exomului clinic (CES). Secvențierea genetică verifică secvența de nucleotide, sau ortografia „literelor”, a genelor pentru a determina dacă există o modificare care afectează funcția genei. Genomul uman este format din 3 milioane de nucleotide, dar numai 1-2% dintre acestea, numite exoni, sunt traduse într-un produs proteic funcțional. Restul de ADN necodificator intercalat între exoni, care nu este tradus, se numește introni (74). CES examinează aproape toți exonii cunoscuți din cele aproximativ 20 000 de gene din genomul uman, care reprezintă o minoritate din materialul genetic al cromozomilor, dar care sunt cel mai probabil să conțină variante cauzatoare de boală (patogene). CES poate include, de asemenea, secvențierea ADN mitocondrial (ADNmt), care interoghează ADN circular mic, extranuclear, situat în mitocondrie, care este moștenit exclusiv pe cale maternă.
Rezultatele posibile pentru CES includ variante pozitive, negative și variante cu semnificație necunoscută. Un rezultat pozitiv înseamnă că sunt identificate variantele cunoscute care cauzează boala (adică patogene), după care pot fi discutate diagnosticul, istoria naturală, prognosticul, riscul de recurență și opțiunile de tratament. Un rezultat negativ înseamnă că nu a fost identificată nicio variantă patogenă detectabilă. Variante cu semnificație necunoscută (alias VUS) înseamnă că, deși au fost identificate modificări genetice, nu există suficiente informații despre modificarea genetică specifică pentru a ști cu certitudine dacă aceasta este cauzatoare de boală. Este de așteptat să existe variații în ADN-ul fiecărui individ, astfel încât testarea concomitentă a probelor parentale pentru comparație poate ajuta la interpretarea laboratorului și clinică a rezultatelor. Randamentul diagnostic al CES este estimat la 30-35% și crește în timp, pe măsură ce descoperirea de gene și cunoștințele despre genomul uman continuă să progreseze (75-77). CES este din ce în ce mai des comandat ca fiind testul genetic larg de primă linie de alegere, având în vedere timpul său de execuție rapid și costul relativ scăzut pentru cantitatea de informații genetice analizate. Limitările CES includ lipsa unei sensibilități de 100%, incapacitatea de a detecta anumite tipuri de modificări genetice (de exemplu, deleții, duplicații, repetări trinucleotidice, mutații intronice profunde sau defecte de metilare) și faptul că un diagnostic poate să nu ofere informații suplimentare despre boală sau să schimbe managementul.
În raportarea CES, variantele patogene detectate întâmplător în genele asociate cu afecțiuni genetice bine cunoscute pot fi raportate ca rezultate secundare (78). Această listă de boli recomandate este curatoriată de Colegiul American de Genetică Medicală (ACMG). Legea privind nediscriminarea informațiilor genetice (Genetic Information Nondiscrimination Act – GINA) este un considerent important atunci când se decide dacă se optează sau nu pentru cunoașterea rezultatelor accidentale (79). GINA protejează indivizii împotriva utilizării abuzive a informațiilor genetice în cadrul asigurărilor de sănătate și al angajărilor, dar nu și al asigurărilor de viață. GINA protejează următoarele informații genetice: istoricul medical al familiei, testele de purtător, testele genetice prenatale, testele de susceptibilitate și predictive și analiza tumorilor sau alte evaluări de gene, mutații sau modificări cromozomiale.
Management
Managementul CDG depinde în mare măsură de simptomele specifice ale individului. Simptomele recurente la pacienții cu CDG includ eșecul de creștere, întârzierea globală a dezvoltării, vărsături, episoade asemănătoare unui accident vascular cerebral și anomalii scheletice. Coagulopatia clinică sau subclinică, endocrinopatia, hepatopatia și defectele cardiace sunt, de asemenea, frecvent întâlnite. Se recomandă testele de laborator de bază pentru a stabili amploarea bolii și monitorizarea de rutină, în special pentru PMM2-CDG. Acestea includ testele funcției hepatice, albumina serică, testele funcției tiroidiene, inclusiv T4 liber, proteina C, proteina S, antitrombina III, factorul IX, analiza de urină și gonadotropinele serice și hormonul de creștere.
Imagistica recomandată include ecocardiografia, ecografia renală, vârsta osoasă, examenul oftalmologic pentru evaluarea cristalinului, a retinei, a mobilității oculare și a presiunii intraoculare. Cu excepția cazului în care se indică altfel, vaccinările de rutină sunt recomandate pentru adulții și copiii afectați de CDG. Titrurile de anticorpi trebuie obținute după vaccinare, deoarece pacienții pot avea un răspuns imunogen suboptimal. Reîncărcarea profilactică a factorilor de coagulare înainte de orice procedură chirurgicală poate fi necesară dacă există deficiențe la momentul inițial.
Evaluarea genetică clinică trebuie întreprinsă pentru a discuta aspectele ereditare ale CDG, precum și pentru a stabili un domiciliu medical pentru acești pacienți complecși. Casa medicală este, de obicei, serviciul de genetică biochimică, deși departamentele de genetică, neurodezvoltare sau neurologie au servit, de asemenea, în această calitate dacă nu este disponibil un serviciu de genetică biochimică dedicat. Adesea sunt necesare trimiteri de specialitate pentru gastroenterologie, hematologie, endocrinologie, suport nutrițional, logopedie, terapie ocupațională, fizică și de hrănire, ortopedie și medicină de reabilitare.
Terapii țintite și prognostic
Tratamentul pentru majoritatea tipurilor de CDG este în mare parte de susținere, cu câteva excepții. MPI-CDG este cel mai eficient tratabil dintre toate tipurile de CDG. Mannoza orală este transformată în mannoză-6-fosfat de către hexokinazele intracelulare, ocolind astfel blocajul enzimatic și producând substratul deficitar. Suplimentarea cu mannoză începe de obicei cu 1 g/kg de greutate corporală pe zi, împărțită în 4-6 doze pe zi. În timp ce enteropatia de pierdere a proteinelor, care poate pune în pericol viața, răspunde în mod special la tratamentul cu manoză, boala hepatică în cazul MPI-CDG poate continua să progreseze. Simptomele clinice se ameliorează rapid, iar CDT de transferrină se normalizează în decurs de câteva luni, deși boala hepatică poate continua să progreseze cu tratamentul (45,80,81).
Cuviința trebuie să fie exercitată în suplimentarea mannozei în timpul sarcinii, deoarece administrarea de mannoză la modele de șoareci hipomorfi hipomorfi cu fosfomanonoza izomerază gravidă a dus la letalitate embrionară și orbire la puii acestora (82). În plus, mannoza intravenoasă a fost asociată cu scăderea stării de conștiență și convulsii, care s-au rezolvat cu administrarea de glucoză (83).
Tratamentul pentru PMM2-CDG este în mare parte de susținere și se bazează pe simptomatologie. Cu toate acestea, în prezent sunt în curs de dezvoltare viitoarele studii clinice privind terapia de înlocuire a substratului mannoză-1-fosfat.
Pentru alte CDG, au fost investigate diverse zaharuri simple orale cu scopul de a îmbunătăți teoretic hipoglucozilarea. Fucose a fost încercată pentru SLC35C1-CDG și galactoza pentru PGM1-CDG și SLC35A2-CDG cu rezultate mixte (84). S-a demonstrat că D-galactoza la 1,0-2,5 g/kg/zi (maximum 50 de grame) îmbunătățește hipoglicemia, coagulopatia și endocrinopatria în PGM1-CDG (85,86). S-a demonstrat, de asemenea, că galactoza îmbunătățește endocrenopatia și coagulopatia la TMEM165-CDG (87) și SLC39A8-CDG. O îmbunătățire clinică considerabilă a fost, de asemenea, raportată la pacienții SLC39A8-CDG cu 15-20 mg/kg/zi de MnSO4 (88). Sunt în curs de desfășurare studii clinice pentru a investiga utilitatea N-acetilmannosaminei (ManNAc) în GNE-CDG (89), iar mai multe studii preclinice sunt în curs de desfășurare pentru alte CDG (90).
În ciuda progreselor medicale, există o mortalitate semnificativă pentru copiii cu CDG în primul an de viață din cauza insuficienței multiorganice sau a infecțiilor severe (91). Sugarii cu CDG pot prezenta o boală multiorganică fulminantă, convulsii intratabile sau hipoalbuminemie severă care evoluează spre anasarhă. Unii pacienți răspund la diureza agresivă și la înlocuirea albuminei, în timp ce alții sunt refractari la tratament. S-a demonstrat că butiratul de sodiu îmbunătățește controlul convulsiilor în CAD-CDG și PIGM-CDG (92). S-a demonstrat, de asemenea, că dieta ketogenică scade frecvența convulsiilor în unele cazuri de PIGA-CDG (93). În timpul episoadelor asemănătoare accidentelor vasculare cerebrale, hidratarea intravenoasă și menținerea unei glicemii normale pot fi utile în timp ce se exclude etiologia vasculară subiacentă trombotică sau hemoragică.
Cu apariția tehnicilor de editare a genomului și o mai bună înțelegere a mecanismului bolilor cuprinse în umbrela diagnostică a CDG, viitorul dezvoltării terapiei țintite rămâne promițător.
Recunoștințe
Am dori să le mulțumim lui Lynne Wolfe, ARNP și Donna Krasnewich, MD, PhD, pentru că ne-au pus la dispoziție fotografii clinice obținute în cadrul studiului Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). De asemenea, am dori să îi mulțumim lui Jenny Thies, MS, LGC pentru expertiza sa în domeniul consilierii genetice.
Finanțare: IJ Chang este susținut de National Institutes of Health T32GM007454.
Nota de subsol
Conflicte de interese: Autorii nu au conflicte de interese de declarat.
Consimțământ informat: A fost obținut consimțământul în cunoștință de cauză în scris de la pacienți pentru publicarea acestui manuscris și a tuturor imaginilor care îl însoțesc.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Tulburări congenitale de glicozilare: o familie de boli care se extinde rapid. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutații în PMM2 care cauzează tulburări congenitale de glicozilare, tip Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Tulburări congenitale de glicozilare: o actualizare a defectelor care afectează biosinteza oligozaharidelor legate de dolicoli. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Lipsa echilibrului Hardy-Weinberg pentru cea mai răspândită mutație PMM2 în CDG-Ia (tulburări congenitale de glicozilare de tip Ia). Jurnalul European de Genetică Umană 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Transferrină serică și lichid cefalorahidian cu deficit de acid sialic într-un sindrom genetic nou recunoscut. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. Nomenclatura CDG: este timpul pentru o schimbare! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Rezolvarea tulburărilor de glicozilare: abordările fundamentale dezvăluie căi complicate. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Ce este nou în CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Congenital disorders of glycosylation. Partea I. Defecte de N-glicozilare a proteinelor. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Constatări clinice, de laborator și moleculare și date de urmărire pe termen lung la 96 de pacienți francezi cu PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) și revizuirea literaturii. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG în Africa de Sud: Descrierea genotip-fenotip a cinci pacienți noi. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Diferite glicoziltransferaze sunt procesate diferențiat pentru secreție, dimerizare și autoglicozilare. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. În: Glicemia: Mora-Montes H. Editor. Glycans: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Manifestări cutanate în CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF system in children with congenital disorders of glycosylation. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. Un spectru larg de prezentări clinice în tulburările congenitale de glicozilare I: o serie de 26 de cazuri. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Creșterea prepubertară în tulburarea congenitală de glicozilare de tip Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Caracteristicile clinice și biochimice ale tulburării congenitale de glicozilare de tip Ic, primul defect recunoscut al reticulului endoplasmatic în sinteza N-glicanului. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Tulburări congenitale de glicozilare: revizuire a bazelor lor moleculare, prezentări clinice și terapii specifice. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1- o boală metabolică ereditară recent descoperită. Manifestări organice multiple, incidență 1/80.000, dificil de tratat. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Ischemie miocardică tranzitorie severă cauzată de cardiomiopatie hipertrofică la un pacient cu tulburare congenitală de glicozilare de tip Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Defecte cardiace conotruncale la trei pacienți cu tulburare congenitală de glicozilare de tip Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Acumularea de lichid pericardic și abdominal în tulburarea congenitală de glicozilare de tip Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Caracteristici clinice la adulții cu tulburări congenitale de glicozilare de tip Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Sindromul glicoproteinei cu deficit de carbohidrați. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modification of the coat protein of the potyvirus Plum pox virus enhances viral infection. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Scandinavian CDG-Ia patients: genotype/phenotype correlation and geographic origin of founder mutations. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 de pacienți adulți francezi cu PMM2 – tulburare congenitală de glicozilare: rezultatul fenotipului pediatric clasic și descrierea unui fenotip cu debut tardiv. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Caracteristicile retiniene ale tulburării congenitale de glicozilare PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Tulburarea congenitală de glicozilare de tip Ia (CDG-Ia): spectrul fenotipic al genotipului R141H/F119L. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Thyroid function in PMM2-CDG: diagnostic approach and proposed management. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Tulburare congenitală de glicozilare de tip Ia care se prezintă ca ataxie cerebeloasă cu debut precoce la un adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Tulburare congenitală de glicozilare de tip 1a: trei frați cu un fenotip neurologic ușor. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Noi tulburări în metabolismul carbohidraților: tulburări congenitale de glicozilare și impactul lor asupra sistemului endocrin. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Tulburare congenitală de glicozilare de tip Ia: eterogenitate în prezentarea clinică de la insuficiență multiviscerală la hipoglicemie hiperinsulinemică ca simptome principale la trei sugari cu deficit de fosfomannomutază. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinologia sindromului glicoproteinei cu deficit de carbohidrați tip 1 de la naștere până la adolescență. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Funcția gonadică și hormonii glicoproteinei în sindromul glicoproteinei cu deficit de carbohidrați (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Spectrul clinic al deficitului de fosfomanoză izomerază, cu o evaluare a tratamentului cu manoză pentru CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Complicații cardiace ale tulburărilor congenitale de glicozilare (CDG): o revizuire sistematică a literaturii. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Hidrops fetalis neimună recurentă asociată cu sindromul glicoproteinei cu deficit de carbohidrați. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with carbohydrate-deficient glycoprotein presentation with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Deficitul de fosfomanoză izomerază și terapia cu manoză. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Hipoglicemie severă ca simptom de prezentare a sindromului glicoproteinei cu deficit de carbohidrați. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hipoglicemie hiperinsulinemică ca semn de prezentare în deficitul de fosfomanoză izomerază: O nouă manifestare a sindromului glicoproteinei cu deficit de carbohidrați tratabilă cu manoză. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Cardul genetic de utilitate clinică pentru: ALG6 tulburare congenitală defectuoasă de glicozilare. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: De la producție la secreție. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Rezultatul clinic pe termen lung și variabilitatea fenotipică în calcinoza tumorală familială hiperfosfatemică și sindromul de hiperostoză hiperfosfatemică cauzată de o nouă mutație GALNT3; raport de caz și revizuire a literaturii. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparan sulfat în dezvoltarea, creșterea și patologia scheletului: cazul exostozelor multiple ereditare. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Extinderea spectrului clinic al deficienței B4GALT7: mutația homozigotă p.R270C cu efect fondator provoacă sindromul Larsen din Insula Reunion. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Diverse afecțiuni musculo-scheletice non-inflamatorii. Calcinoză tumorală familială hiperfosfatemică (FGF23, GALNT3 și αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Ablația genei Galnt3 duce la concentrații scăzute ale factorului de creștere a fibroblastelor 23 (Fgf23) intact în circulație și la hiperfosfatemie, în ciuda unei expresii crescute a Fgf23. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. O mutație într-o enzimă de biosinteză a gangliozidelor, ST3GAL5, are ca rezultat sindromul sare & piper, o tulburare neurocutanată cu glicozilarea glicolipidică și glicoproteică alterată. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutațiile în B4GALNT1 (GM2 sintetază) stau la baza unei noi tulburări de biosinteză a gangliozidelor. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. Mutațiile PIGA cauzează encefalopatii epileptice cu debut precoce și caracteristici distinctive. Neurologie. Neurologie 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Mutația hipomorfă a promotorului în PIGM cauzează deficiența ereditară de glicozilfosfatidilinositol. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Mutațiile hipomorfe în PGAP2, care codifică o proteină de remodelare a ancorei GPI, cauzează dizabilitate intelectuală autosomal-recesivă. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. Fenotipul unei mutații germinale în PIGA: gena mutantă somatic în hemoglobinuria paroxistică nocturnă. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutațiile în PIGO, un membru al căii de sinteză a ancorei GPI, cauzează hiperfosfatasie cu retard mental. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. Un nou sindrom de dizabilitate intelectuală cauzat de deficiența ancorei GPI din cauza mutațiilor homozigote în PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutațiile în gena glicozilfosfatidilinositol PIGL cauzează sindromul CHIME. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal heemoglobinuria (PNH) este cauzată de mutații somatice în gena PIG-A. EMBO J 1994;13:110-7.
- Brodsky RA. Hemoglobinuria paroxistică nocturnă. Sânge 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Curente în etica contemporană. GINA, ADA și discriminarea genetică la angajare. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Trombo-embolism recurent la un copil cu o tulburare congenitală de glicozilare (CDG) de tip Ib și tratament cu mannoză. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Dezvoltarea bolii hepatice în ciuda tratamentului cu mannoză la doi pacienți cu CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Suplimentele de manoză induc letalitate embrionară și orbire la șoarecii hipomorfi hipomorfi cu izomeroză de fosfomanoză. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Convulsii și stupoare în timpul terapiei cu manoză intravenoasă la un pacient cu sindrom CDG tip 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Suplimentarea cu glucoză în deficitul de aderență a leucocitelor de tip II. Sânge 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Deficiența moștenită de glicozilfosfatidil inositol: o CDG tratabilă. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Suplimentarea orală cu D-galactoză în PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Suplimentarea cu galactoză la pacienții cu TMEM165-CDG salvează defectele de glicozilare. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. Deficiența SLC39A8: corecție biochimică și îmbunătățire clinică majoră prin terapia cu mangan. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Terapii de monosacaride orale pentru a inversa hiposialilarea renală și musculară într-un model de șoarece de miopatie GNE. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside (De la bancă la patul de spital). Int J Mol Sci 2018;19:1304.
- Krasnewich D. Tulburări de glicozilare umană. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Terapie țintită pentru deficiența GPI moștenită. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev 2016;38:848-51.