- Overview
- Epidemiologia
- Klasyfikacja biochemiczna i nomenklatura
- Genetyka
- Patofizjologia
- Uszkodzenia glikozylacji białek N
- Uszkodzenia glikozylacji O-linked i połączone defekty glikozylacji N- i O-linked
- Wady biosyntezy glikozylacji lipidów i kotwic GPI
- Objawy kliniczne
- Uszkodzenia glikozylacji białek N-linked
- PMM2-CDG (CDG-Ia, niedobór PMM2)
- MPI-CDG (CDG-Ib, niedobór izomerazy mannofosforanowej)
- ALG6-CDG (niedobór glukozylotransferazy 1)
- Uszkodzenia glikozylacji O-linked i połączone defekty glikozylacji N- i O-linked
- Uszkodzenia glikozylacji lipidów i biosyntezy kotwic GPI
- Diagnostyka
- Uszkodzenia związane z glikozylacją białek N
- Uszkodzenia glikozylacji O-linked i połączone defekty glikozylacji N- i O-linked
- Uszkodzenia glikozylacji lipidów i biosyntezy kotwic GPI
- Analiza molekularna
- Zarządzanie
- Terapie celowane i rokowanie
- Podziękowania
- Przypis
Overview
Glikozylacja jest procesem dodawania reszt cukrowych do białek i lipidów w różnych szlakach komórkowych. Wrodzone zaburzenia glikozylacji (CDG) są genetycznie i klinicznie heterogenną grupą ponad stu chorób spowodowanych defektami na różnych etapach szlaków syntezy lub modyfikacji glikanów. Większość z tych monogenowych chorób jest dziedziczona w sposób autosomalny recesywny, ale opisano również formy autosomalne dominujące i sprzężone z chromosomem X.
CDG zazwyczaj objawiają się wieloukładowo, najczęściej opóźnieniem rozwoju, brakiem przyrostu masy ciała, hipotonią, zaburzeniami neurologicznymi, hepatopatią i koagulopatią. U osób dotkniętych chorobą mogą również występować choroby oczu, skóry i serca, jak również dysmorfia twarzy. Chociaż zmiany neurologiczne i opóźnienia poznawcze są obserwowane u większości chorych, istnieją przypadki, a nawet typy, w których nie występują objawy neurologiczne. Biorąc pod uwagę szeroką etiologię kliniczną i genetyczną CDG, diagnostyka kliniczna opiera się na wysokim indeksie podejrzliwości w chorobie wieloukładowej.
Analiza transferyny z niedoborem węglowodanów w surowicy (CDT) jest badaniem przesiewowym pierwszego rzutu u pacjentów z podejrzeniem CDG, ale jest ograniczona w wykrywaniu do defektów N-glikozylacji z niedoborem kwasu sialowego. Kolejne badania obejmują analizę glikanów dolicholowych i badania genetyczne. Wczesne rozpoznanie tej grupy gwałtownie rosnących chorób jest bardzo ważne, ponieważ niektóre CDG są uleczalne. Leczenie defektów glikozylacji jest głównie wspomagające, chociaż dostępne są terapie celowane dla MPI-CDG, SLC35C1-CDG, PIGM-CDG i PGM1-CDG. Szczegóły dotyczące tych terapii znajdują się w części „Terapie celowane i rokowanie” poniżej. Niniejszy przegląd koncentruje się na najczęstszych typach CDG z rozpoznawalnymi fenotypami lub metodami leczenia, a jego odbiorcami są lekarze podstawowej opieki zdrowotnej.
Epidemiologia
Zachorowalność i rozpowszechnienie wszystkich typów CDG łącznie nie zostały dobrze ustalone, chociaż pacjenci są zgłaszani na całym świecie z prawie każdego tła etnicznego, a obie płcie są w równym stopniu dotknięte chorobą. Szacowana częstość występowania w populacjach europejskich i afroamerykańskich wynosi 1/10,000 w oparciu o częstość nosicielstwa znanych patogennych wariantów w 53 genach (1-4). Częstość występowania najczęściej rozpoznawanej CDG, PMM2-CDG, waha się od 1/20,000 w populacjach holenderskich do 1/77,000 w Estonii na podstawie pojedynczych doniesień (5,6). Do tej pory zgłoszono mniej niż 100 przypadków dla większości typów CDG.
Klasyfikacja biochemiczna i nomenklatura
Ogólnie, CDG są obecnie sklasyfikowane w czterech kategoriach-(I) glikozylacja N-związana, (II) glikozylacja O-związana, (III) połączona N- i O-związana/wielokrotna glikozylacja oraz (IV) defekty biosyntezy kotwic lipidowych i glikozylofosfatydyloinozytolu (GPI).
Wada N-liniowej glikozylacji białek PMM2-CDG, wcześniej znana jako CDG typu Ia, była pierwszą CDG zgłoszoną przez Jaekena w 1980 roku i pozostaje zdecydowanie najczęstszą CDG do dnia dzisiejszego (7). PMM2-CDG został początkowo nazwany „zespołem glikoprotein z niedoborem węglowodanów” ze względu na liczne nieprawidłowości glikoprotein w surowicy, które można zaobserwować podczas ogniskowania izoelektrycznego transferyny w surowicy u osób dotkniętych chorobą. Historycznie, CDG były klasyfikowane na podstawie wzorców analizy izoform transferyny – wzorce typu I były przypisywane do defektów montażu i transferu glikanów związanych z dolicholem, zlokalizowanych w cytoplazmie lub ER, a wzorce typu II były przypisywane do defektów przetwarzania w aparacie Golgiego. Z tego punktu gałązki, CDG zostały następnie nazwane alfabetycznie w kolejności odkrycia.
Wraz z pojawieniem się powszechnej diagnostyki molekularnej, nomenklatura CDG została zaktualizowana w 2008 roku w celu określenia molekularnej etiologii choroby, odzwierciedlając gwałtowny wzrost szlaków i zaburzeń, które nie pasują do wcześniej ustalonych dychotomicznych kategorii. Obecnie, nomenklatura CDG jest oznaczana przez nazwę dotkniętego genu (nieocynkowana, nazwy genów na www.genenames.org), a następnie przez -CDG (np. PMM2-CDG) (8).
Genetyka
Ogromna większość wrodzonych zaburzeń glikozylacji jest dziedziczona w sposób autosomalny recesywny, z jedną mutacją dziedziczoną od każdego bezobjawowego (nosiciela) rodzica. Badania molekularne, zwykle z zastosowaniem metod sekwencjonowania nowej generacji, są niezbędne do ustalenia rozpoznania genetycznego. Badanie rodziców na obecność znanego wariantu może potwierdzić dziedziczenie lub wystąpienie de novo. W przypadku dziedziczenia autosomalnego recesywnego ryzyko nawrotu u rodzeństwa i w każdej ciąży osoby dotkniętej chorobą wynosi 25% w przypadku bycia dotkniętym chorobą, 50% w przypadku bycia bezobjawowym nosicielem i 25% w przypadku bycia niedotkniętym chorobą.
Kilka przypadków CDG ma dziedziczenie autosomalne dominujące (N-linked: GANAB-CDG, PRKCSH-CDG; O-linked: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Mniej jest sprzężonych z X (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). Większość dominujących i niektóre X-linked form CDG są spowodowane mutacjami de novo. Konkretne choroby i geny są opisane poniżej w części „patofizjologia”.
Dane o mutacjach dla wszystkich opublikowanych genów dla CDG są dostępne w Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Informacje o specyficznych wariantach genów są dostępne w Leiden Open Variation Database ze zintegrowanymi narzędziami patogenności in silico (http://www.lovd.nl/3.0/home). Streszczenia kliniczne dotyczące konkretnych genów można znaleźć w internetowym serwisie Mendelian Inheritance in Man (http://www.omim.org/) lub w bardziej ograniczonym zakresie w GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Biorąc pod uwagę małą liczbę dotkniętych pacjentów w przypadku większości podtypów CDG, korelacja genotyp-fenotyp jest trudna do ustalenia.
Patofizjologia
Do tej pory opisano ponad 130 typów CDG (9,10). Ze względu na wszechobecność szlaków glikozylacji, CDG są niezwykle zróżnicowane w swojej biochemicznej patogenezie. Liczne białka i lipidy (tj. sfingolipidy i glikolipidy) ulegają glikozylacji z monosacharydami i/lub oligosacharydami, określanymi wspólnym mianem glikanów, w różnych przedziałach komórkowych. Ich lokalizacja subkomórkowa jest zróżnicowana, ale większość defektów występuje w obrębie ER lub aparatu Golgiego. Charakterystyka kliniczna i etiologia genetyczna najczęstszych CDG według szlaków jest podsumowana w Tabeli S1.
Pośród białek, glikany są opisane przez ich powiązanie z łańcuchem polipeptydowym-N-glikany są dołączone do grupy amidowej asparaginy (Asn), podczas gdy O-glikany są dołączone do grupy hydroksylowej seryny lub treoniny. Synteza N-glikanów wymaga stopniowej budowy cukrów połączonych nukleotydami w cytozolu, łączenia w retikulum endoplazmatycznym i przetwarzania w aparacie Golgiego. W przeciwieństwie do tego, synteza O-glikanów wymaga montażu, ale nie przetwarzania, dlatego defekty O-glikozylacji występują głównie w aparacie Golgiego.
Uszkodzenia glikozylacji białek N
N-glikozylacja obejmuje kowalencyjne przyłączenie struktur węglowodanowych do grupy amidowej łańcucha bocznego reszt Asn w ramach konsensusowego miejsca akceptorowego Asn-X-Ser/Thr, translokację polipeptydu substratowego do retikulum endoplazmatycznego w celu przebudowy i dalszej modyfikacji łańcucha N-glikanu w aparacie Golgiego (11,12). Defekty w dowolnym miejscu na szlaku syntezy, montażu i przetwarzania mogą prowadzić do choroby klinicznej.
PMM2-CDG jest powodowana przez patogenne warianty w genie fosfomannomutazy 2 (PMM2), prowadzące do niedoboru enzymu PMM2, który katalizuje cytozolową konwersję mannozy-6-fosforanu do mannozy-1-fosforanu w drugim etapie syntezy mannozy z difosforanu guanozyny (GDP). Większość pacjentów posiada złożone heterozygotyczne patogenne mutacje typu missense (www.lovd.nl/PMM2). Najczęstszy powtarzający się patogenny wariant p.Arg141His występuje u około 40% dotkniętych chorobą osób pochodzenia europejskiego, a p.Phe119Leu jest również często spotykany w północnej Europie (1). Opisano korelacje genotyp-fenotyp dla PMM2-CDG (3,13,14).
MPI-CDG jest zaburzeniem autosomalnym recesywnym spowodowanym przez patogenne warianty w genie izomerazy fosforanu mannozy (MPI) prowadzące do niedoboru izomerazy fosfomannozy (MPI). MPI normalnie katalizuje pierwszy etap syntezy GDP-mannozy (tj. konwersję fruktozo-6-fosforanu do mannozy-6-fosforanu), ale fruktozo-6-fosforan nie gromadzi się wewnątrzkomórkowo, ponieważ może być również metabolizowany przez szlak glikolityczny. Dlatego MPI-CDG, choć biochemicznie podobny do PMM2-CDG, nie powoduje tak znacznego zajęcia neurologicznego i wieloukładowego. CDT jest również testem przesiewowym z wyboru dla MPI-CDG, który wykazuje wzorzec typu 1. Diagnoza może być następnie potwierdzona molekularnie lub przez aktywność MPI fibroblastów/leukocytów.
ALG6-CDG jest chorobą recesywną spowodowaną mutacjami w ALG6, prowadzącymi do nieprawidłowego przyłączenia trzech cząsteczek glukozy do pośrednich cząsteczek mannozy związanych z dolicholem i do hipoglikozylacji glikoprotein surowicy (15).
Uszkodzenia glikozylacji O-linked i połączone defekty glikozylacji N- i O-linked
O-glikozylacja polega na stopniowym dodawaniu łańcuchów węglowodanowych do reszt serynowych, treoninowych i hydroksylizynowych białek przez glikozylotransferazy w aparacie Golgiego (16). Kilka typów O-linked glikanów zostało powiązanych z chorobami człowieka, nazwanych przez pierwszy cukier dołączony do reszty aminokwasowej (17).
Wady biosyntezy glikozylacji lipidów i kotwic GPI
Kotwice GPI są glikolipidami, które przechodzą sekwencyjny montaż w retikulum endoplazmatycznym i modyfikacje w obrębie Golgiego. Zaburzenia biosyntezy kotwic GPI spowodowane niedoborem enzymów są nazwane alfabetycznie według kolejności odkrycia, a nie chronologicznie według etapu montażu. Po zsyntetyzowaniu, kotwice GPI rezydują na błonach plazmatycznych i wiążą setki białek powierzchni komórkowej, pełniąc wiele funkcji komórkowych. Większość z tych chorób jest dziedziczona autosomalnie recesywnie, z godnym uwagi wyjątkiem niedoboru PIGA sprzężonego z chromosomem X.
Objawy kliniczne
Zważywszy na wszechobecność szlaków glikozylacji, praktycznie każdy układ narządowy może być zaangażowany w CDG, chociaż większość przypadków obejmuje nieprawidłowości neurologiczne. Niektóre CDG występują z ichtiozą, w tym MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (rysunek 1) i PIGL-CDG (18,19). Prawie wszystkie CDG objawiają się chorobą wieloukładową w ciągu pierwszych kilku lat życia, z wyjątkiem niektórych, które dotyczą tylko jednego układu narządowego (np, siatkówka w DHDDS-CDG; złącze nerwowo-mięśniowe w ALG2-CDG, ALG14-CDG, CFPT1-CDG; mózg w ST3GAL3-CDG, TUSC3-CDG; skóra lub mięśnie szkieletowe w POGLUT1-CDG, POFUT1-CDG; chrząstka w EXT1/EXT2-CDG; wątroba w TMEM199-CDG; krwinki czerwone w SEC23B-CDG). Wiek wystąpienia i ciężkość choroby może wahać się od śmierci noworodka do prawie bezobjawowej dorosłości, a także wszelkie permutacje pomiędzy. Najczęściej zgłaszana konstelacja objawów obejmuje opóźnienie rozwoju, brak przyrostu masy ciała, hipotonię, nieprawidłowości neurologiczne, hipoglikemię i zmienne zaburzenia wątroby, oczu, skóry, przewodu pokarmowego, immunologiczne, szkieletowe i krzepnięcia (19).
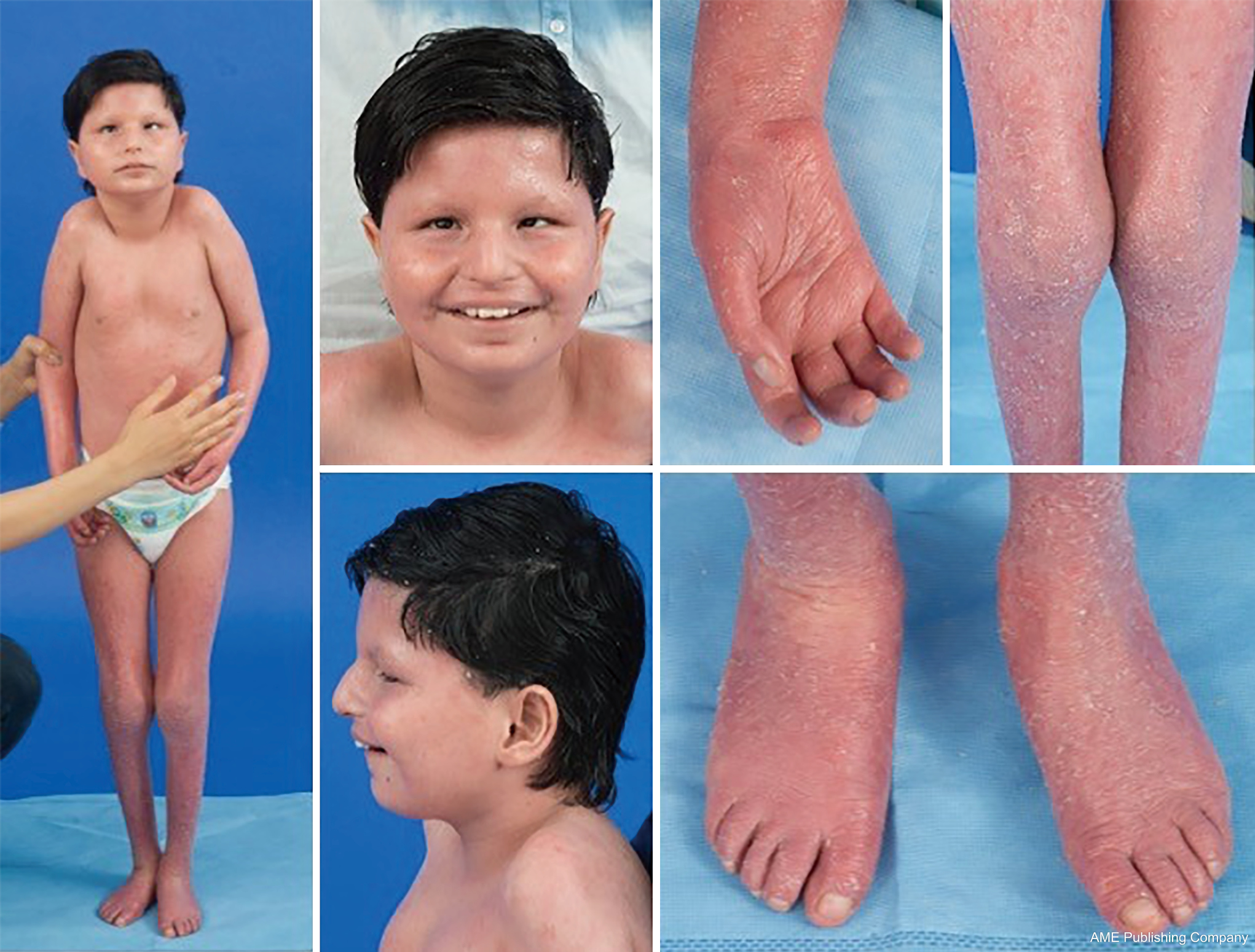
Kompletny fenotyp dla wielu podtypów CDG pozostaje do pełnego określenia ze względu na rzadkość zgłaszanych przypadków. Dlatego CDG powinno być rozważane w każdym przypadku choroby wieloukładowej, szczególnie w przypadkach z komponentem neurologicznym lub niespecyficznym opóźnieniem rozwoju o niejasnej etiologii.
Pomimo, że patofizjologia wielu objawów pozostaje do wyjaśnienia, związek pomiędzy niektórymi szlakami glikozylacji a specyficznymi objawami klinicznymi został wyjaśniony. Na przykład, niepowodzenie w rozwoju obserwowane w wielu typach CDG można przypisać hipoglikozylacji i upośledzonemu tworzeniu kilku glikoprotein w obrębie ścieżki wzrostu insuliny, w tym IGF-1, ALS i IGFBP-3 (20). W miarę jak coraz lepiej rozumiemy tę grupę złożonych zaburzeń, CDG są coraz częściej rozpoznawane u osób z nieuchwytnymi diagnozami. Zajęcie poszczególnych narządów przez różne CDG podsumowano w Tabeli S1. Poniżej omówimy cechy kliniczne najczęstszych form i form z ukierunkowanym leczeniem CDG.
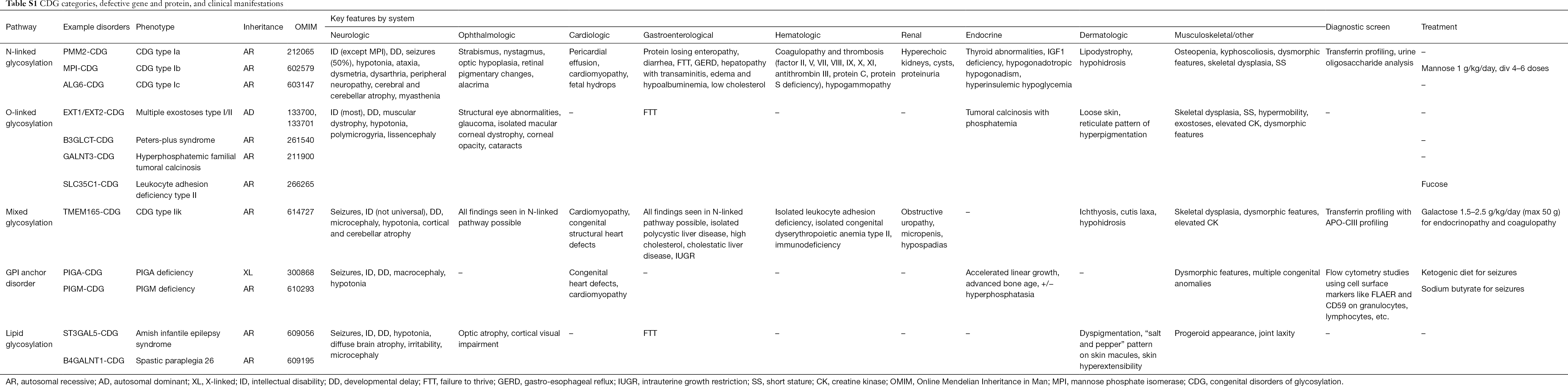
Pełna tabela
Uszkodzenia glikozylacji białek N-linked
Jako najczęściej diagnozowane CDG, fenotyp zaburzeń glikozylacji N-linked jest często uznawany za klasyczny. Jednakże spektrum fenotypowe CDG jest dość zróżnicowane, a wiele CDG może nie dawać stereotypowych objawów związanych z PMM2-CDG.
PMM2-CDG (CDG-Ia, niedobór PMM2)
PMM2-CDG jest najczęstszą CDG, z ponad 700 zgłoszonymi przypadkami na całym świecie. Charakteryzuje się wielosystemową ciężką chorobą w wieku niemowlęcym, chorobą neurologiczną i opóźnieniem rozwoju w dzieciństwie i/lub stabilną niepełnosprawnością intelektualną w wieku dorosłym (21,22).
W niemowlęctwie, PMM2-CDG prezentuje się z nieprawidłowościami neurologicznymi typowo krótko po urodzeniu, a mianowicie zezem i nieprawidłowymi ruchami gałek ocznych, hipoplazją móżdżku, hipotonią, opóźnieniem psychoruchowym, ataksją, hipotonią i hiporefleksją. U niemowląt może również wystąpić choroba wątroby, zespół nerczycowy i torbiele nerek, wysięk osierdziowy i kardiomiopatia przerostowa, brak zdolności do wzrostu i niewydolność wielonarządowa prowadząca do zgonu w ciągu pierwszego roku życia nawet u 20% chorych (21,23-28).
U pacjentów z PMM2-CDG opisano konstelację cech dysmorficznych (ryc. 2,3). Obejmują one hipoplastyczny móżdżek, dysmorfię twarzy (np. duże, dysplastyczne uszy), odwrócone brodawki sutkowe i nieprawidłowe rozmieszczenie tkanki tłuszczowej na pośladkach lub w okolicy nadłonowej, które może ustępować z wiekiem (14,21,29-32). Opisywano, że pacjenci mają otwarte i radosne usposobienie. Objawy są bardzo zróżnicowane, chociaż zeza można zaobserwować u ponad 70% pacjentów (21,23,33-35). Odwrócone sutki i nieprawidłowe poduszeczki tłuszczowe obserwuje się u około 25-50% pacjentów (36).

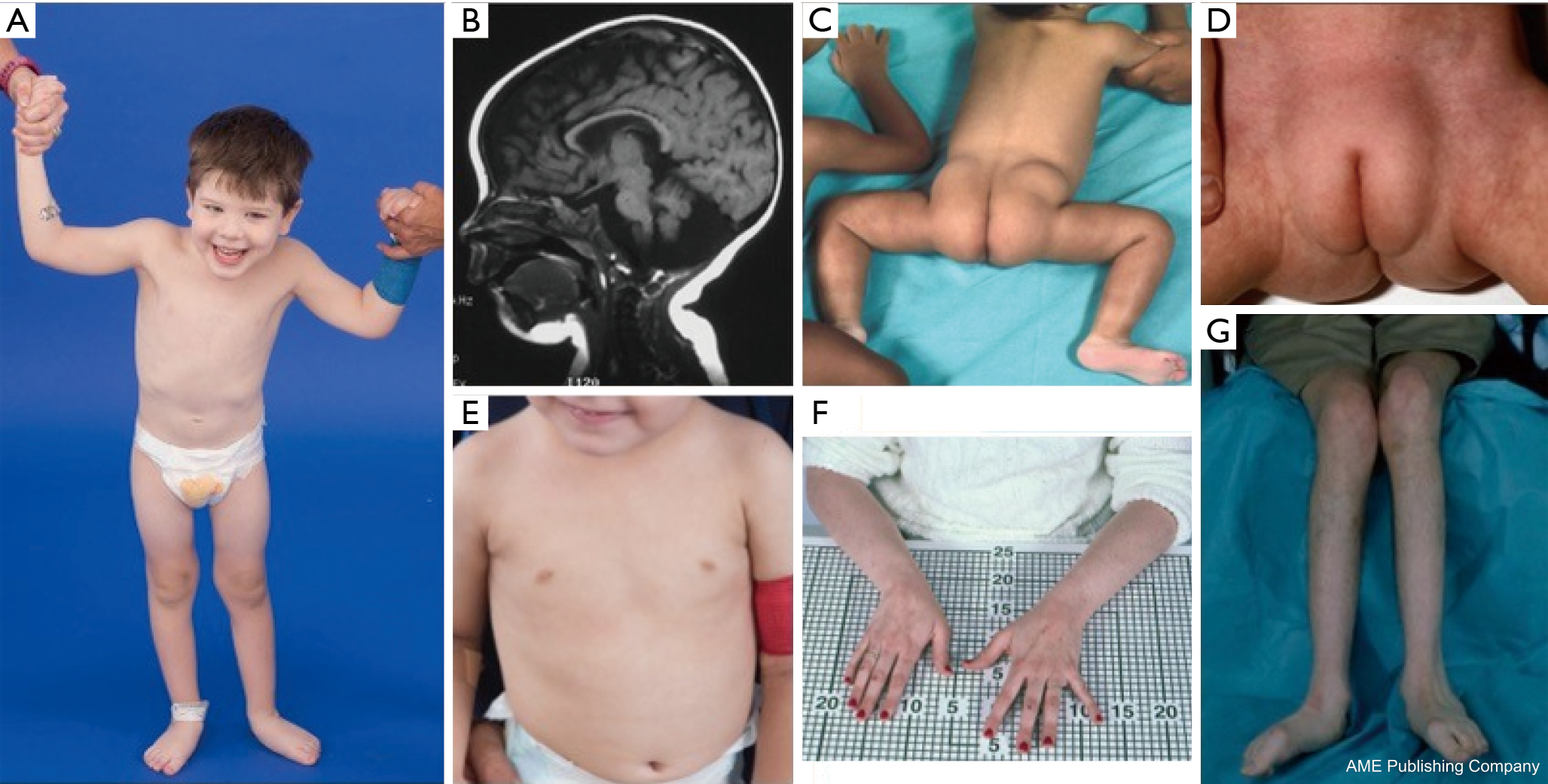
W dzieciństwie, dotknięte osoby mogą rozwijać pigmentozę siatkówki, epizody podobne do udaru i napady, opóźnienia mowy i ruchu oraz neuropatię obwodową. Konstytucjonalnie, pacjenci często nie rozwijają się z powodu zaburzeń odżywiania i zaburzeń żołądkowo-jelitowych oraz globalnych opóźnień rozwojowych. Można zaobserwować podwyższone wartości transaminaz wątrobowych bez konsekwencji klinicznych, które zazwyczaj normalizują się do 5 roku życia z okazjonalnymi wahaniami w czasie choroby (21,24). Biopsja wątroby jest rzadko wskazana w CDG, chyba że podejrzewa się zwłóknienie wątroby (1). Kliniczna niedoczynność tarczycy jest rzadka, ale u pacjentów z CDG należy oznaczyć poziom hormonów tarczycy i wolnej T4, co może wykazać niski poziom globuliny wiążącej tarczycę (TBG) i przejściowe podwyższenie poziomu hormonu stymulującego tarczycę (TSH) (37). Wady rozwojowe wątroby i dróg żółciowych nie były zgłaszane u pacjentów z PMM2-CDG.
Dorośli z PMM2-CDG mogą dożyć 7. lub 8. dekady życia ze stabilnym opóźnieniem poznawczym, neuropatią obwodową oraz postępującą kifoskoliozą piersiową i kręgosłupa z osteopenią lub osteoporozą (34). Coraz częściej rozpoznawanym objawem jest ataksja móżdżkowa wraz z zajęciem wielonarządowym (38-40). Zaburzenia endokrynologiczne obejmują hiperprolaktynemię, uwalnianie hormonu wzrostu z hiperglikemią, insulinooporność i hipoglikemię hiperinsulinemiczną (41,42). U kobiet dotkniętych tą chorobą hipogonadyzm hipogonadotropowy może prowadzić do braku wtórnego rozwoju płciowego lub braku jajników (41,43,44). Pacjenci mogą być narażeni na zwiększone ryzyko zakrzepicy z powodu obniżonego stężenia czynników krzepnięcia, w tym czynników IV, IX i XI, antytrombiny III, białka C i białka S (29).
MPI-CDG (CDG-Ib, niedobór izomerazy mannofosforanowej)
MPI-CDG jest unikalny, ponieważ pacjenci dotknięci tą chorobą mają niewielkie lub żadne objawy neurologiczne, a niektóre objawy choroby są uleczalne przez doustne podawanie mannozy (2). Objawy są głównie wątrobowo-jelitowe, bez cech dysmorficznych lub opóźnień poznawczych. Pacjenci zazwyczaj prezentują nawracające wymioty, znaczną hipoglikemię, brak przyrostu masy ciała, potencjalnie zagrażającą życiu enteropatię z utratą białka, zmiany zwłóknieniowe wątroby i poszerzenie dróg żółciowych (45-51). Pacjenci są narażeni na zwiększone ryzyko zdarzeń zakrzepowych z powodu niskiego stężenia w surowicy białka C i S oraz antytrombiny III.
ALG6-CDG (niedobór glukozylotransferazy 1)
ALG6-CDG jest drugim co do częstości występowania defektem N-glikozylacji charakteryzującym się podobnym, ale łagodniejszym fenotypem niż PMM2-CDG. Pacjenci z ALG6-CDG nie rozwijają się, mają opóźnienia w rozwoju, hipotonię, drgawki, zeza, ataksję, koagulopatię i dysmorfię twarzy (np. nisko osadzone uszy, hiperteloryzm i makroglosję). Podobnie jak w przypadku MPI-CDG, mogą oni również mieć enteropatię tracącą białko. Dodatkowo, pacjenci dotknięci tą chorobą mogą mieć nieprawidłowości w budowie szkieletu, w tym brachycefalię, zniekształcenia palców i skoliozę. Pacjenci dotknięci tą chorobą zazwyczaj nie mają zapalenia barwnikowego siatkówki ani hipoplazji móżdżku (52).
Uszkodzenia glikozylacji O-linked i połączone defekty glikozylacji N- i O-linked
Z uwagi na znaczną obecność O-glikanów w białkach zawierających mucynę, w tym glikozaminoglikanów (GAG) i powierzchni nabłonka (53), zaburzenia syntezy GAG zazwyczaj prowadzą do dysplazji szkieletowych lub chorób tkanki łącznej. U pacjentów dotkniętych tą chorobą mogą występować nieprawidłowości mięśniowo-szkieletowe, skórne i stawowe (np. wiotkość stawów, liczne egzostozy, chrzęstniakomięsaki), a także objawy neurologiczne (54-56). Na przykład, N-acetylogalaktozaminylotransferaza 3 (GALNT3) O-glikozyluje hormon fosfaturyczny, FGF23, zapobiegając rozszczepieniu proteolitycznemu i umożliwiając jego nienaruszone wydzielanie. Niedobór GALNT3 prowadzi do rodzinnej kalcynozy guzowatej, charakteryzującej się hiperfosfatemią i ektopowymi zwapnieniami (57,58).
Uszkodzenia glikozylacji lipidów i biosyntezy kotwic GPI
Glikosfingolipidy i ich sialilowane pochodne, gangliozydy, ulegają ekspresji głównie przez neurony. Defekty w rozkładzie gangliozydów prowadzą do ich akumulacji i dobrze scharakteryzowanych lizosomalnych chorób spichrzeniowych. Z drugiej strony, defekty w biosyntezie gangliozydów, takie jak ST3GAL5-CDG i B4GALNT1-CDG są niezwykle rzadkie i prowadzą do ciężkiej choroby neurodegeneracyjnej. Pacjenci mogą występować z paraplegią spastyczną, poważnym opóźnieniem intelektualnym, padaczką i objawami nieneurologicznymi, w tym dysplazją szkieletową, cechami dysmorficznymi i nieprawidłową pigmentacją skóry (59,60).
Mutacje w wielu genach w obrębie szlaku biosyntezy kotwicy GPI powodują różnorodne liczne anomalie wrodzone, niepełnosprawność intelektualną i padaczkę. Najlepiej scharakteryzowany defekt biosyntezy GPI, X-liniowy niedobór PIGA, objawia się skurczami u niemowląt z hiperrytmią, hipotonią, licznymi nieprawidłowościami mózgu i dysmorfią twarzy. Pacjenci mogą również mieć zmienne choroby skóry, wątroby, serca i nerek (61-69). Niektóre mutacje w obrębie PIGA powodują fenotypowo odrębną chorobę napadową nocną hemoglobinurię (PNH), nabyte zaburzenie niewydolności szpiku kostnego (70,71).
Diagnostyka
Gdy klinicznie podejrzewa się CDG, pierwszym krokiem jest zlecenie badań biochemicznych CDG w osoczu lub surowicy, w tym badań CDT i N-glikanów. Analiza CDT i N-glikanów w surowicy może wykryć tylko defekty N-glikozylacji, dlatego nie będą one przydatne w różnicowaniu izolowanych defektów O-glikozylacji lub kotwicy GPI. Analiza izoform transferyny została pierwotnie uzyskana przez ogniskowanie izoelektryczne transferyny, ponieważ brak syntezy N-glikanów powoduje częściowy niedobór kwasu sialowego, który zmienia ładunek na transferynie surowicy, a następnie jej katodową migrację na polu elektroforetycznym. Jednak analiza transferryny i N-glikanów oparta na spektrometrii masowej w dużym stopniu zastąpiła obecnie ogniskowanie izoelektryczne, identyfikując specyficzne zmiany w oligosacharydach za pomocą masy i ładunku (72).
Uszkodzenia związane z glikozylacją białek N
Wyniki CDT transferryny w surowicy są podawane jako stosunek mono-oligosacharydów/di-oligosacharydów transferryny, a-oligosacharydów/di-oligosacharydów transferryny, tri-sialo/di-oligosacharydów transferryny, apolipoproteiny CIII-1/apolipoproteiny CIII-2 oraz apolipoproteiny CIII-0/apolipoproteiny CIII-2. Te ilościowe wyniki będą również pochodzić z interpretacją wzoru ustaleń.
Typ I wzór transferyny CDT charakteryzuje się zwiększonymi pasmami di- i asialotransferyny i wskazuje na defekty w syntezie N-glikanów w cytosolu lub retikulum endoplazmatycznym. Wzorzec typu II charakteryzuje się zwiększonymi pasmami di- i asialotransferyny oraz pasmami tri- i/lub monosialotransferyny i wskazuje na defekty w przetwarzaniu N-glikanów w aparacie Golgiego (73).
Jeśli wykryto wzorzec CDT transferyny w surowicy typu I, niedobór PMM2 lub niedobór MPI powinien być na czele różnicowania, ponieważ PMM2-CDG jest najczęstszą CDG, a MPI-CDG jest uleczalna i potencjalnie śmiertelna, jeśli nie jest leczona. Aby zróżnicować rozpoznania, należy wykonać profilowanie N-glikanów, sekwencjonowanie molekularne lub badania enzymatyczne. Rozpoznanie PMM2-CDG lub MPI-CDG jest potwierdzane przez badania molekularne wykazujące bialleliczne patogenne warianty w PMM2 lub MPI, a następnie aktywność enzymu PMM lub MPI w leukocytach lub fibroblastach, jeśli patogenność wariantów genetycznych jest niepewna. Analiza N-glikanów lub analiza molekularna pozwoliłaby odróżnić większość ALG-CDG od PMM2 lub MPI-CDG (15).
Wzór CDT transferyny w surowicy typu II wskazuje na defekty Golgiego, takie jak niedobór N-acetyloglukozaminotransferazy (GnT) II (CDG typu IIA, MGAT2-CDG). Analiza izoformy apolipoproteiny CIII (Apo-CIII) jest badaniem uzupełniającym dla profilu CDT typu II, ponieważ mierzy defekty glikozylacji mucyny typu O w aparacie Golgiego. Istnieje ograniczona czułość dla CDT lub Apo-CIII w wykrywaniu CDG typu II. Dlatego profilowanie N-glikanów i O-glikanów oraz panel molekularny lub sekwencjonowanie eksomu powinny być wykonane, gdy te testy kliniczne są dostępne. Wzorce glikozylacji transferyny mogą normalizować się sporadycznie; dlatego powtórzenie badań może być wskazane u pacjentów z wysokim indeksem podejrzenia. Wyniki fałszywie dodatnie mogą być uzyskiwane u pacjentów z ostrym kryzysem dziedzicznej nietolerancji fruktozy, galaktozemią, ostrą chorobą wątroby i niektórymi infekcjami bakteryjnymi. Żaden z biochemicznych testów CDG nie jest w stanie wykryć wszystkich CDG, dlatego nawet w przypadku prawidłowych wyników badań przesiewowych, przy dużym podejrzeniu klinicznym można wykonać badanie panelu genów molekularnych lub sekwencjonowanie eksomu. I odwrotnie, biochemiczne i funkcjonalne potwierdzenie wyników badań molekularnych jest również niezbędne, ponieważ większość pacjentów z CDG jest nosicielami co najmniej jednej łagodnej i często nowej mutacji typu missense.
Uszkodzenia glikozylacji O-linked i połączone defekty glikozylacji N- i O-linked
Diagnostyka opiera się na sekwencjonowaniu molekularnym, ponieważ analiza izoform transferyny nie wykryje izolowanych defektów O-glikozylacji. Połączone defekty glikozylacji N- i O-linked mogą być wykryte przez CDT, analizę ApoCIII oraz analizę N-glikanów i O-glikanów w osoczu.
Uszkodzenia glikozylacji lipidów i biosyntezy kotwic GPI
Cytometria przepływowa granulocytów krwi mierzy ekspresję na powierzchni komórek białek zakotwiczonych w GPI, takich jak CD16 i CD24. Analiza cytometrii przepływowej krwinek białych lub krwinek czerwonych pod kątem określonych białek powierzchniowych zakotwiczonych w GPI jest dostępna klinicznie jako test na PNH spowodowaną nabytymi mutacjami w genie PIGA. Badanie PNH może ujawnić nieprawidłowości w innych niedoborach kotwic GPI, ale diagnoza opiera się głównie na analizie molekularnej.
Analiza molekularna
Największą wydajność diagnostyczną w przypadku CDG zapewnia panel sekwencjonowania genów nowej generacji lub sekwencjonowanie egzomu klinicznego (CES). Sekwencjonowanie genetyczne sprawdza sekwencję nukleotydów, czyli pisownię liter, genów w celu określenia, czy istnieje zmiana, która wpływa na funkcję genu. Genom ludzki składa się z 3 milionów nukleotydów, ale tylko 1-2% z nich, zwanych eksonami, jest tłumaczonych na funkcjonalny produkt białkowy. Pozostały niekodujący DNA, znajdujący się pomiędzy eksonami, który nie ulega translacji, nazywany jest intronami (74). CES bada prawie wszystkie znane eksony z około 20 000 genów w ludzkim genomie, które stanowią mniejszość materiału genetycznego w chromosomach, ale są najbardziej prawdopodobne, że zawierają warianty powodujące choroby (patogenne). CES może również obejmować sekwencjonowanie mitochondrialnego DNA (mtDNA), które bada małe, pozajądrowe, koliste DNA zlokalizowane w mitochondriach, które jest dziedziczone wyłącznie po matce.
Możliwe wyniki dla CES obejmują wynik pozytywny, negatywny i warianty o nieznanym znaczeniu. Wynik pozytywny oznacza, że zidentyfikowano znane warianty powodujące chorobę (tj. patogenne), po czym można omówić diagnozę, historię naturalną, rokowanie, ryzyko nawrotu i opcje leczenia. Wynik negatywny oznacza, że nie zidentyfikowano wykrywalnych wariantów patogennych. Warianty o nieznanym znaczeniu (VUS) oznaczają, że chociaż zidentyfikowano zmiany genetyczne, nie ma wystarczających informacji na temat konkretnej zmiany genetycznej, aby ostatecznie stwierdzić, czy jest ona przyczyną choroby. Spodziewane są różnice w DNA każdej osoby, dlatego równoczesne badanie próbek rodziców w celu porównania może pomóc w laboratoryjnej i klinicznej interpretacji wyników. Wydajność diagnostyczną CES szacuje się na 30-35% i wzrasta ona z czasem, w miarę odkrywania genów i postępu wiedzy o ludzkim genomie (75-77). CES jest coraz częściej zlecane jako badanie genetyczne pierwszego wyboru ze względu na szybki czas realizacji i niski koszt względny w stosunku do ilości analizowanych informacji genetycznych. Ograniczenia CES obejmują brak 100% czułości, niezdolność do wykrywania pewnych typów zmian genetycznych (np. delecji, duplikacji, powtórzeń trinukleotydowych, głębokich mutacji intronowych lub defektów metylacji) oraz fakt, że diagnoza może nie dostarczyć dodatkowych informacji o chorobie lub zmienić sposób postępowania.
W raportowaniu CES przypadkowo wykryte patogenne warianty w genach związanych z dobrze znanymi schorzeniami genetycznymi mogą być zgłaszane jako wyniki wtórne (78). Ta lista zalecanych chorób została opracowana przez American College of Medical Genetics (ACMG). Ustawa o niedyskryminacji w zakresie informacji genetycznej (Genetic Information Nondiscrimination Act, GINA) jest istotnym czynnikiem przy podejmowaniu decyzji, czy zdecydować się na uczenie się przypadkowych wyników badań, czy też nie (79). GINA chroni osoby przed niewłaściwym wykorzystaniem informacji genetycznych w ubezpieczeniach zdrowotnych i zatrudnieniu, ale nie w ubezpieczeniach na życie. GINA chroni następujące informacje genetyczne: wywiad medyczny dotyczący rodziny, badania na nosicielstwo, genetyczne badania prenatalne, badania podatności i badania predykcyjne oraz analiza guzów lub inne oceny genów, mutacji lub zmian chromosomalnych.
Zarządzanie
Zarządzanie CDG zależy w dużej mierze od specyficznych objawów występujących u danej osoby. Powtarzające się objawy u pacjentów z CDG obejmują brak przyrostu masy ciała, globalne opóźnienie rozwoju, wymioty, epizody podobne do udaru mózgu oraz nieprawidłowości w układzie kostnym. Często obserwuje się również kliniczną lub podkliniczną koagulopatię, endokrynopatię, hepatopatię i wady serca. Zaleca się wykonanie podstawowych badań laboratoryjnych w celu określenia stopnia zaawansowania choroby oraz rutynowe monitorowanie, szczególnie w przypadku PMM2-CDG. Obejmują one testy czynnościowe wątroby, albuminę w surowicy, testy czynnościowe tarczycy, w tym wolną T4, białko C, białko S, antytrombinę III, czynnik IX, badanie moczu oraz gonadotropiny i hormon wzrostu w surowicy.
Zalecane badania obrazowe obejmują echokardiogram, USG nerek, wiek kostny, badanie okulistyczne w celu oceny soczewki, siatkówki, ruchomości oka i ciśnienia wewnątrzgałkowego. Jeśli nie wskazano inaczej, u dorosłych i dzieci chorych na CDG zaleca się rutynowe szczepienia. Po szczepieniu należy oznaczyć miano przeciwciał, ponieważ u pacjentów może wystąpić suboptymalna odpowiedź immunogenna. Profilaktyczne uzupełnianie czynników krzepnięcia przed jakimikolwiek zabiegami chirurgicznymi może być konieczne, jeśli niedobory występują na poziomie wyjściowym.
Należy przeprowadzić ocenę genetyki klinicznej w celu omówienia dziedzicznych aspektów CDG, jak również ustanowienia domu medycznego dla tych złożonych pacjentów. Domem medycznym jest zwykle dział genetyki biochemicznej, chociaż działy genetyki, neurorozwoju lub neurologii również pełniły tę funkcję, jeśli dedykowany dział genetyki biochemicznej jest niedostępny. Często konieczne jest skierowanie do specjalisty z zakresu gastroenterologii, hematologii, endokrynologii, dietetyki, logopedii, terapii zajęciowej, fizycznej i żywienia, ortopedii i rehabilitacji.
Terapie celowane i rokowanie
Leczenie większości typów CDG jest w dużej mierze wspomagające, z kilkoma wyjątkami. MPI-CDG jest najskuteczniej uleczalną spośród wszystkich CDG. Mannoza podawana doustnie jest przekształcana do mannozy-6-fosforanu przez wewnątrzkomórkowe heksokinazy, dzięki czemu omijany jest blok enzymatyczny i wytwarzany jest niedoborowy substrat. Suplementację mannozą rozpoczyna się zwykle od 1 g/kg masy ciała na dobę, podzieloną na 4-6 dawek dziennie. Podczas gdy potencjalnie zagrażająca życiu enteropatia z utratą białka szczególnie dobrze reaguje na leczenie mannozą, choroba wątroby w MPI-CDG może postępować. Objawy kliniczne ulegają szybkiej poprawie, a transferyna CDT normalizuje się w ciągu kilku miesięcy, chociaż choroba wątroby może nadal postępować w trakcie leczenia (45,80,81).
Należy zachować ostrożność podczas uzupełniania mannozy w czasie ciąży, ponieważ podawanie mannozy w ciężarnych hipomorficznych modelach mysich izomerazy fosfomannozy powodowało śmiertelność zarodków i ślepotę u ich szczeniąt (82). Ponadto, dożylne podanie mannozy było związane z obniżeniem świadomości i drgawkami, które ustąpiły po podaniu glukozy (83).
Leczenie PMM2-CDG jest w dużej mierze wspomagające i oparte na symptomatologii. W przypadku innych CDG badano różne doustne cukry proste w celu teoretycznej poprawy hipoglikozylacji. Fukoza została wypróbowana dla SLC35C1-CDG, a galaktoza dla PGM1-CDG i SLC35A2-CDG z mieszanymi wynikami (84). Wykazano, że D-galaktoza w dawce 1,0-2,5 g/kg/dzień (maksymalnie 50 gramów) poprawia hipoglikemię, koagulopatię i endokrynopatię w PGM1-CDG (85,86). Wykazano również, że galaktoza poprawia endokrynopatię i koagulopatię w TMEM165-CDG (87) i SLC39A8-CDG. Znaczną poprawę kliniczną odnotowano również u pacjentów z SLC39A8-CDG otrzymujących MnSO4 w dawce 15-20 mg/kg/dobę (88). Trwają badania kliniczne mające na celu zbadanie przydatności N-acetylomannozy (ManNAc) w GNE-CDG (89), a kilka badań przedklinicznych jest w trakcie realizacji dla innych CDG (90).
Mimo postępów w medycynie, u dzieci z CDG występuje znaczna śmiertelność w ciągu pierwszego roku życia z powodu niewydolności wielonarządowej lub ciężkiej infekcji (91). Niemowlęta z CDG mogą występować z piorunującą chorobą wielonarządową, trudnymi do opanowania drgawkami lub ciężką hipoalbuminemią postępującą do anasarca. Niektórzy pacjenci reagują na agresywną diurezę i substytucję albuminami, podczas gdy inni są oporni na leczenie. Wykazano, że maślan sodu poprawia kontrolę napadów w CAD-CDG i PIGM-CDG (92). Wykazano również, że dieta ketogenna zmniejsza częstość napadów w niektórych przypadkach PIGA-CDG (93). Podczas epizodów podobnych do udaru mózgu, dożylne nawodnienie i utrzymanie prawidłowego stężenia glukozy we krwi może być pomocne, podczas gdy podstawowa naczyniowa etiologia zakrzepowa lub krwotoczna jest wykluczona.
Wraz z pojawieniem się technik edycji genomu i lepszym zrozumieniem mechanizmu chorób objętych diagnostycznym parasolem CDG, przyszłość rozwoju terapii celowanej pozostaje obiecująca.
Podziękowania
Chcielibyśmy podziękować Lynne Wolfe, ARNP i Donnie Krasnewich, MD, PhD za udostępnienie nam zdjęć klinicznych uzyskanych w ramach badania Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). Chcielibyśmy również podziękować Jenny Thies, MS, LGC za jej doświadczenie w doradztwie genetycznym.
Finansowanie: IJ Chang jest wspierany przez National Institutes of Health T32GM007454.
Przypis
Konflikty interesów: Autorzy nie mają konfliktu interesów do zgłoszenia.
Informed Consent: Pisemna świadoma zgoda została uzyskana od pacjentów na publikację tego manuskryptu i wszelkich towarzyszących obrazów.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG nomenklatura: czas na zmianę! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Congenital disorders of glycosylation. Część I. Defekty N-glikozylacji białek. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Wyniki kliniczne, laboratoryjne i molekularne oraz dane z długoterminowej obserwacji u 96 francuskich pacjentów z PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) i przegląd literatury. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG in South Africa: Genotype-Phenotype Description of Five Novel Patients. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Different glycosyltransferases are differentially processed for secretion, dimerization, and autoglycosylation. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In: Mora-Montes H. Editor. Glycans: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Skin manifestations in CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF system in children with congenital disorders of glycosylation. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertal growth in congenital disorder of glycosylation type Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N-glycan synthesis. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–a recently discovered hereditary metabolic disease. Objawy wielonarządowe, częstość występowania 1/80,000, trudna do leczenia. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Carbohydrate-deficient glycoprotein syndrome. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modification of the coat protein of the potyvirus Plum pox virus enhances viral infection. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Scandinavian CDG-Ia patients: genotype/phenotype correlation and geographic origin of founder mutations. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 francuskich dorosłych pacjentów z PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Thyroid function in PMM2-CDG: diagnostic approach and proposed management. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. New disorders in carbohydrate metabolism: congenital disorders of glycosylation and their impact on the endocrine system. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadal function and glycoprotein hormones in the carbohydrate-deficient glycoprotein (CDG) syndrome. Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Zespół glikoproteinowy z niedoborem węglowodanów typu Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hipoglikemia hiperinsulinemiczna jako objaw prezentujący w niedoborze izomerazy fosfomannozy: A new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG6 defective congenital disorder of glycosylation. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: From production to secretion. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparan sulfate in skeletal development, growth, and pathology: the case of hereditary multiple exostoses. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Miscellaneous non-inflammatory musculoskeletal conditions. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Ablacja genu Galnt3 prowadzi do niskich stężeń czynnika wzrostu fibroblastów 23 (Fgf23) i hiperfosfatemii pomimo zwiększonej ekspresji Fgf23. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J 1994;13:110-7.
- Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Nurty we współczesnej etyce. GINA, ADA i dyskryminacja genetyczna w zatrudnieniu. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Recurrent thrombo-embolism in a child with a congenital disorder of glycosylation (CDG) type Ib and treatment with mannose. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Rozwój choroby wątroby pomimo leczenia mannozą u dwóch pacjentów z CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Seizures and stupor during intravenous mannose therapy in a patient with CDG syndrome type 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Fucose supplementation in leukocyte adhesion deficiency type II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Wrodzony niedobór glikozylofosfatydylo inozytolu: uleczalne CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Doustna suplementacja D-galaktozą w PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Galactose Supplementation in Patients With TMEM165-CDG Rescues the Glycosylation Defects. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8 deficiency: biochemiczna korekta i znaczna poprawa kliniczna dzięki terapii manganem. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Zaburzenia glikozylacji u człowieka. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev 2016;38:848-51.
.