- Overzicht
- Epidemiologie
- Biochemische classificatie en nomenclatuur
- Genetica
- Pathofysiologie
- N-gekoppelde eiwitglycosylatiedefecten
- O-linked glycosylatiedefecten en gecombineerde N- en O-linked glycosylatiedefecten
- Lipideglycosylering en GPI-ankerbiosynthesedefecten
- Klinische manifestaties
- N-gekoppelde eiwitglycosylatiedefecten
- PMM2-CDG (CDG-Ia, PMM2-deficiëntie)
- MPI-CDG (CDG-Ib, mannosefosfaat isomerase deficiëntie)
- ALG6-CDG (glucosyltransferase 1 deficiëntie)
- O-linked glycosylation defects and combined N- and O-linked glycosylation defects
- Lipideglycosylerings- en GPI-ankerbiosynthesedefecten
- Diagnose
- N-gekoppelde proteïneglycosyleringsdefecten
- O-linked glycosylatiedefecten en gecombineerde N- en O-linked glycosylatiedefecten
- Lipideglycosylering en GPI-anker biosynthese defecten
- Moleculaire analyse
- Behandeling
- Gerichte therapieën en prognose
- Acknowledgements
- Footnote
Overzicht
Glycosylatie is het proces van het toevoegen van suikerresiduen aan eiwitten en lipiden in verschillende cellulaire trajecten. Congenitale aandoeningen van de glycosylatie (CDG) zijn een genetisch en klinisch heterogene groep van meer dan honderd ziekten die worden veroorzaakt door defecten in verschillende stappen van de glycansynthese of -modificatieroutes. De meeste van deze monogene ziekten erven autosomaal recessief over, maar autosomaal dominante en X-gebonden vormen zijn ook beschreven.
CDG presenteren zich meestal met multi-systemische manifestaties, meestal ontwikkelingsachterstand, falen om te gedijen, hypotonie, neurologische afwijkingen, hepatopathie, en coagulopathie. Ook kunnen oog-, huid- en hartafwijkingen en gelaatsmorfismen optreden. Hoewel neurologische veranderingen en cognitieve achterstand worden gezien in de meerderheid van de getroffen personen, zijn er bepaalde gevallen en zelfs types die geen neurologische manifestaties hebben. Gezien de brede klinische en genetische etiologie van CDG, berust de klinische diagnose op een hoge verdenkingsindex bij multi-systemische ziekte.
Serum carbohydraat deficiënt transferrine (CDT) analyse is de eerstelijns screeningstest bij patiënten met verdenking op CDG, maar is beperkt in detectie tot N-glycosylatiedefecten met siaalzuurdeficiënties. Tests in de volgende lijn omvatten dolichol-gekoppelde glycaananalyse en genetische tests. Vroege diagnose van deze groep van exponentieel groeiende ziekten is belangrijk, omdat sommige CDG behandelbaar zijn. De behandeling van glycosyleringsdefecten is hoofdzakelijk ondersteunend, hoewel gerichte therapieën beschikbaar zijn voor MPI-CDG, SLC35C1-CDG, PIGM-CDG, en PGM1-CDG. Details over deze behandelingen zijn te vinden in het gedeelte “Gerichte therapieën en prognose” hieronder. De focus van dit overzicht zal liggen op de meest voorkomende vormen van CDG met herkenbare fenotypes of behandelingen, met als doelgroep de eerstelijns zorgverleners.
Epidemiologie
De incidentie en prevalentie van alle vormen van CDG samen zijn niet goed vastgesteld, hoewel er wereldwijd patiënten zijn gerapporteerd met bijna elke etnische achtergrond en beide geslachten in gelijke mate worden getroffen. De geschatte prevalentie in Europese en Afro-Amerikaanse populaties is 1/10.000 op basis van dragerschapsfrequenties van bekende pathogene varianten in 53 genen (1-4). De prevalentie van de meest gediagnosticeerde CDG, PMM2-CDG, varieert van 1/20,000 in Nederlandse populaties en 1/77,000 in Estland op basis van geïsoleerde rapporten (5,6). Tot op heden zijn voor de meeste CDG-typen minder dan 100 gevallen gemeld.
Biochemische classificatie en nomenclatuur
In het algemeen worden CDG momenteel ingedeeld in vier categorieën-(I) N-gekoppelde glycosylering, (II) O-gekoppelde glycosylering, (III) gecombineerde N- en O-gekoppelde/meervoudige glycosylering, en (IV) defecten in de biosynthese van lipiden en glycosylphosphatidylinositol (GPI) ankers.
Het N-gekoppelde eiwitglycosyleringsdefect PMM2-CDG, voorheen bekend als CDG type Ia, was het eerste CDG dat door Jaeken in 1980 werd gerapporteerd en blijft tot op heden verreweg het meest voorkomende CDG (7). PMM2-CDG kreeg aanvankelijk de naam “carbohydraat-deficiënt glycoproteïne syndroom” wegens meervoudige serum glycoproteïne afwijkingen gezien door isoelectrische focussering van serum transferrine bij getroffen personen. In het verleden werden CDG geclassificeerd volgens patronen van transferrine isovorm analyse – type I patronen werden toegeschreven aan dolichol-gekoppelde glycaan assemblage en transfer defecten die gelokaliseerd werden in het cytoplasma of ER, en type II patronen werden toegeschreven aan processing defecten in het Golgi apparaat. Met de komst van wijdverspreide moleculaire diagnostiek werd de CDG-nomenclatuur in 2008 bijgewerkt om de moleculaire etiologie van de ziekte te specificeren, als gevolg van de exponentiële groei van pathways en aandoeningen die niet netjes in de eerder vastgestelde dichotome categorieën pasten. Momenteel wordt de CDG-nomenclatuur aangeduid door de naam van het getroffen gen (niet-geitaliseerd, gennamen op www.genenames.org), gevolgd door -CDG (bv. PMM2-CDG) (8).
Genetica
De overgrote meerderheid van aangeboren glycosylatiestoornissen erft op een autosomaal recessieve manier over, waarbij één mutatie wordt geërfd van elke asymptomatische (drager) ouder. Moleculaire tests, meestal met sequencingmethoden van de volgende generatie, zijn nodig om een genetische diagnose te stellen. Ouderschapsonderzoek voor de bekende variant kan overerving versus de novo optreden bevestigen. Bij autosomaal recessieve overerving is het recidiverisico voor broers en zussen en elke zwangerschap van een aangedaan individu 25% voor het aangedaan zijn, 50% voor het asymptomatisch drager zijn, en 25% voor het niet aangedaan zijn.
Een handvol CDG heeft een autosomaal dominante overerving (N-gebonden: GANAB-CDG, PRKCSH-CDG; O-gebonden: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Minder zijn X-gebonden (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). De meeste dominante en sommige X-gebonden vormen van CDG zijn het gevolg van de novo mutaties. Specifieke ziekten en genen worden hieronder beschreven in de sectie “pathofysiologie”.
Mutatiegegevens voor alle gepubliceerde genen voor CDG zijn beschikbaar in de Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Informatie over specifieke genvarianten is beschikbaar in de Leiden Open Variation Database met geïntegreerde in silico pathogeniciteitsgereedschappen (http://www.lovd.nl/3.0/home). Klinische samenvattingen voor specifieke genen kunnen worden gevonden op de Online Mendelian Inheritance in Man (http://www.omim.org/) of in een beperkter bereik op GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Gezien het kleine aantal getroffen patiënten voor de meeste CDG subtypes, is genotype-fenotype correlatie moeilijk vast te stellen.
Pathofysiologie
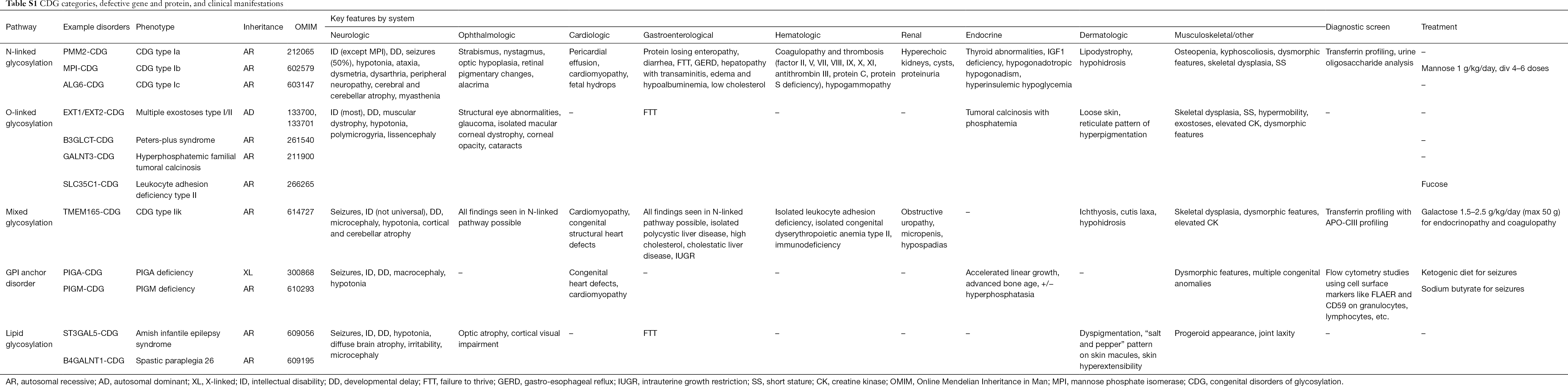
Over 130 soorten CDG zijn tot op heden gerapporteerd (9,10). Gezien de alomtegenwoordigheid van glycosyleringswegen, zijn CDG uiterst divers in hun biochemische pathogenese. Talrijke proteïnen en lipiden (d.w.z. sfingolipiden en glycolipiden) ondergaan glycosylatie met monosachariden en/of oligosachariden, gezamenlijk glycanen genoemd, in verschillende cellulaire compartimenten. Hun subcellulaire locaties zijn divers, maar de meeste defecten treden op binnen het ER of het Golgi-apparaat. Klinische kenmerken en genetische etiologie van meer voorkomende CDG per pathway is samengevat in Tabel S1.
Van de eiwitten worden glycanen beschreven door hun koppeling aan de polypeptideketen-N-glycanen zijn verbonden aan de amidegroep van asparagine (Asn) terwijl O-glycanen verbonden zijn aan de hydroxylgroep van hetzij serine of threonine. De synthese van N-glycanen vereist een stapsgewijze opbouw van nucleotide-gebonden suikers in het cytosol, assemblage in het endoplasmatisch reticulum, en verwerking in het Golgi-apparaat. Daarentegen vereist de O-glycan synthese wel assemblage maar geen verwerking, zodat O-glycosyleringsdefecten voornamelijk in het Golgi-apparaat optreden.
N-gekoppelde eiwitglycosylatiedefecten
N-glycosylatie omvat de covalente binding van koolhydraatstructuren aan de zijketen amidegroep van Asn residuen binnen een consensus Asn-X-Ser/Thr acceptor site, translocatie van het substraatpolypeptide naar het endoplasmatisch reticulum voor remodellering, en verdere modificatie van de N-glycan keten binnen het Golgi (11,12). Defecten in de synthese, assemblage en verwerkingsroute kunnen leiden tot klinische ziekte.
PMM2-CDG wordt veroorzaakt door pathogene varianten in het gen voor fosfomannomutase 2 (PMM2), wat leidt tot een tekort aan het PMM2-enzym dat de cytosolische omzetting van mannose-6-fosfaat naar mannose-1-fosfaat katalyseert in de tweede stap van de mannosesynthese op basis van guanosinedifosfaat (GDP). De meeste patiënten hebben samengestelde heterozygote pathogene missense mutaties (www.lovd.nl/PMM2). De meest voorkomende recurrente pathogene variant p.Arg141His wordt aangetroffen bij ongeveer 40% van de getroffen personen van Europese afstamming, en p.Phe119Leu wordt ook vaak aangetroffen in Noord-Europa (1). Er zijn genotype-fenotype correlaties gerapporteerd voor PMM2-CDG (3,13,14).
MPI-CDG is een autosomaal recessieve aandoening die wordt veroorzaakt door pathogene varianten in het mannose fosfaat isomerase (MPI) gen die leiden tot deficiëntie van fosfomannose isomerase (MPI). MPI katalyseert normaal gesproken de eerste stap van de GDP-mannose synthese (d.w.z. de omzetting van fructose-6-fosfaat in mannose-6-fosfaat), maar fructose-6-fosfaat hoopt zich niet intracellulair op omdat het ook door de glycolytische route kan worden gemetaboliseerd. Daarom veroorzaakt MPI-CDG, hoewel biochemisch vergelijkbaar met PMM2-CDG, niet zo’n significante neurologische en multisystemische betrokkenheid. CDT is ook de screeningstest bij uitstek voor MPI-CDG, dat een type 1-patroon laat zien. De diagnose kan vervolgens moleculair worden bevestigd of door fibroblast/leukocyt MPI activiteit.
ALG6-CDG is een recessieve ziekte veroorzaakt door mutaties in ALG6, leidend tot abnormale binding van drie glucosemoleculen aan dolichol-gekoppelde mannose-tussenproducten en stroomafwaartse hypoglycosylering van serumglycoproteïnen (15).
O-linked glycosylatiedefecten en gecombineerde N- en O-linked glycosylatiedefecten
O-glycosylatie omvat de stapsgewijze toevoeging van koolhydraatketens aan serine-, threonine- en hydroxylysineresten van eiwitten door glycosyltransferases in het Golgi-apparaat (16). Verschillende soorten O-gekoppelde glycanen zijn in verband gebracht met ziekten bij de mens, genoemd naar de eerste suiker die aan het aminozuurresidu is gehecht (17).
Lipideglycosylering en GPI-ankerbiosynthesedefecten
GPI-ankers zijn glycolipiden die opeenvolgende assemblage ondergaan in het endoplasmatisch reticulum en modificaties binnen het Golgi. GPI anker biosynthese stoornissen als gevolg van enzym deficiënties worden genoemd in alfabetische volgorde van ontdekking en niet chronologisch volgens stap van de assemblage. Eenmaal gesynthetiseerd bevinden GPI-ankers zich op plasmamembranen en binden ze honderden eiwitten aan het celoppervlak, waarbij ze een groot aantal cellulaire functies vervullen. De meeste van deze ziekten zijn autosomaal recessief, met als opmerkelijke uitzondering de X-gebonden PIGA-deficiëntie.
Klinische manifestaties
Gezien de alomtegenwoordigheid van glycosyleringswegen kan vrijwel elk orgaansysteem betrokken zijn bij CDG, hoewel de meeste gevallen gepaard gaan met neurologische afwijkingen. Sommige CDG presenteren zich met ichthyosis, waaronder MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (Figuur 1), en PIGL-CDG (18,19). Bijna alle CDG presenteren zich met een multisystemische ziekte binnen de eerste levensjaren, behalve enkele die slechts één enkel orgaansysteem aantasten (d.w.z, netvlies in DHDDS-CDG; neuromusculaire junctie in ALG2-CDG, ALG14-CDG, CFPT1-CDG; hersenen in ST3GAL3-CDG, TUSC3-CDG; huid of skeletspieren in POGLUT1-CDG, POFUT1-CDG; kraakbeen in EXT1/EXT2-CDG; lever in TMEM199-CDG; rode bloedcellen in SEC23B-CDG). De aanvangsleeftijd en de ernst van de ziekte kunnen variëren van neonataal dodelijk tot bijna asymptomatisch op volwassen leeftijd, en elke permutatie daartussen. De meest voorkomende symptomen zijn ontwikkelingsachterstand, gebrek aan doorbloeding, hypotonie, neurologische afwijkingen, hypoglykemie en variabele lever-, oog-, huid-, gastro-intestinale, immunologische, skelet- en stollingsafwijkingen (19).
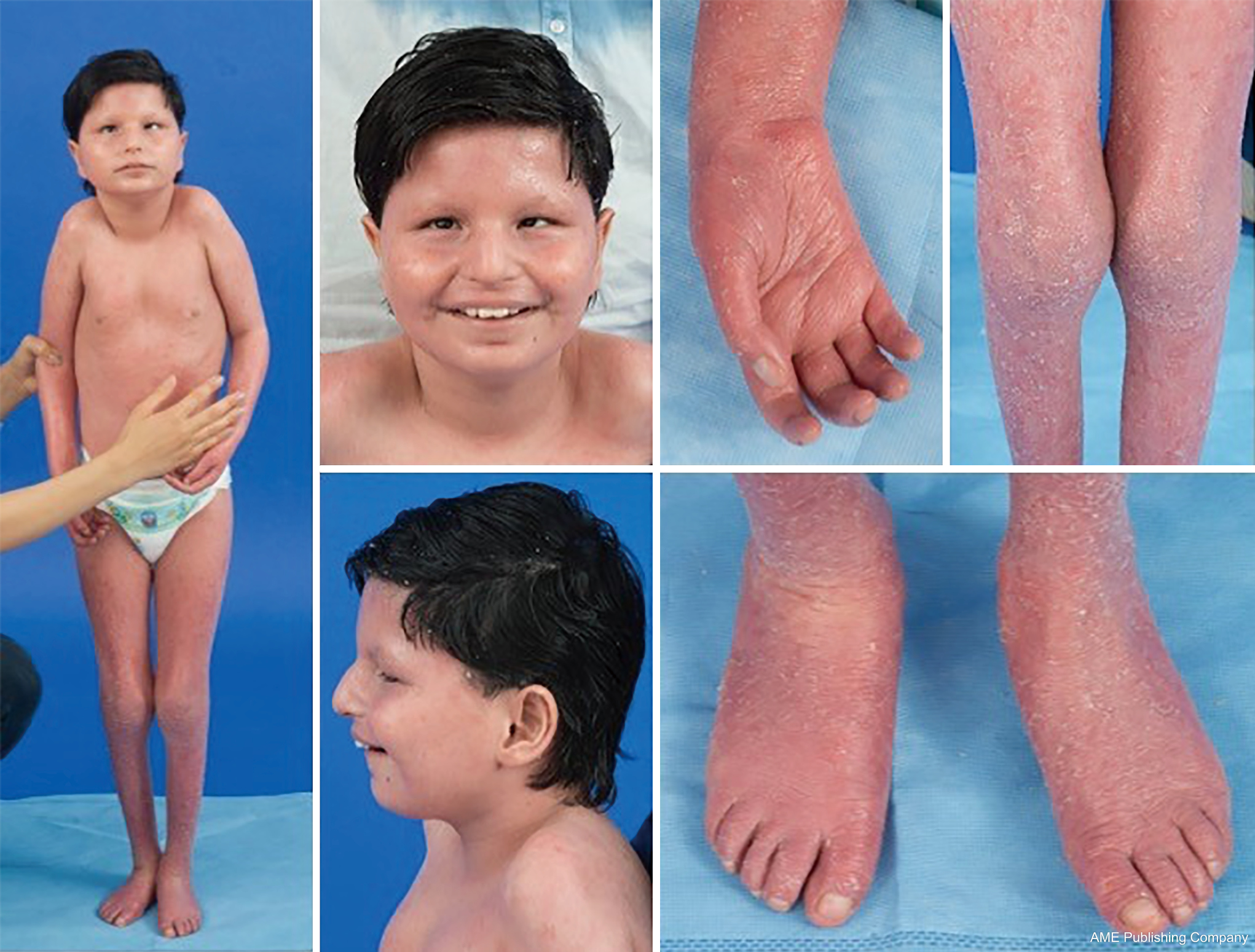
Het volledige fenotype van vele CDG-subtypes moet nog volledig worden afgebakend door de zeldzaamheid van de gerapporteerde gevallen. Daarom moet CDG worden overwogen in elke setting van multi-systemische ziekte, vooral in gevallen met een neurologische component of aspecifieke ontwikkelingsachterstand met onduidelijke etiologie.
Hoewel de pathofysiologie van multitude symptomen nog moet worden opgehelderd, is de relatie tussen bepaalde glycosylatie pathways en specifieke klinische symptomen wel opgehelderd. Bijvoorbeeld, het niet gedijen dat in vele soorten CDG wordt gezien is toe te schrijven aan de hypoglycosylatie en de verminderde vorming van verscheidene glycoproteïnen binnen de insuline groei pathway waaronder IGF-1, ALS, en IGFBP-3 (20). Naarmate we deze groep complexe aandoeningen beter begrijpen, worden CDG in toenemende mate herkend bij personen met ongrijpbare diagnoses. De betrokkenheid van de orgaansystemen van verschillende CDG zijn samengevat in Tabel S1. We zullen de klinische kenmerken van de meest voorkomende vormen en vormen met gerichte behandelingen van CDG hieronder bespreken.
Volledige tabel
N-gekoppelde eiwitglycosylatiedefecten
Als de meest gediagnosticeerde CDG wordt het fenotype van N-gekoppelde glycosylatiestoornissen vaak aangeprezen als de klassieke presentatie. Het fenotypische spectrum van CDG is echter zeer divers, en veel CDG presenteren zich niet met de stereotiepe symptomen die geassocieerd worden met PMM2-CDG.
PMM2-CDG (CDG-Ia, PMM2-deficiëntie)
PMM2-CDG is de meest voorkomende CDG, met meer dan 700 gemelde gevallen wereldwijd. Het wordt gekenmerkt door multisystemische ernstige ziekte in de kindertijd, neurologische ziekte en ontwikkelingsachterstand in de kindertijd, en/of stabiele verstandelijke handicap in de volwassenheid (21,22).
In de kindertijd presenteert PMM2-CDG zich met neurologische afwijkingen die typisch zijn kort na de geboorte, namelijk strabismus en abnormale oogbewegingen, cerebellaire hypoplasie, hypotonie, psychomotorische retardatie, ataxie, hypotonie, en hyporeflexie. Zuigelingen kunnen ook leveraandoeningen, nefrotisch syndroom en niercysten, pericardiale effusie en hypertrofische cardiomyopathie, falen om door te gedijen, en multi-orgaanfalen hebben, resulterend in de dood binnen het eerste levensjaar bij tot 20% van de getroffen individuen (21,23-28).
Een constellatie van dysmorfe kenmerken is beschreven bij patiënten met PMM2-CDG (Figuren 2,3). Deze omvatten een hypoplastisch cerebellum, gezichtsdysmorfismen (d.w.z., grote, dysplastische oren), omgekeerde tepels, en abnormale verdeling van vetweefsel over de billen of suprapubische regio die met de leeftijd kan verdwijnen (14,21,29-32). Van de patiënten wordt gezegd dat ze een vrolijke en vrolijke houding hebben. De presentatie is zeer variabel, hoewel strabismus kan worden gezien bij meer dan 70% van de getroffen patiënten (21,23,33-35). Omgekeerde tepels en abnormale vetkussentjes worden gezien bij ongeveer 25-50% van de patiënten (36).

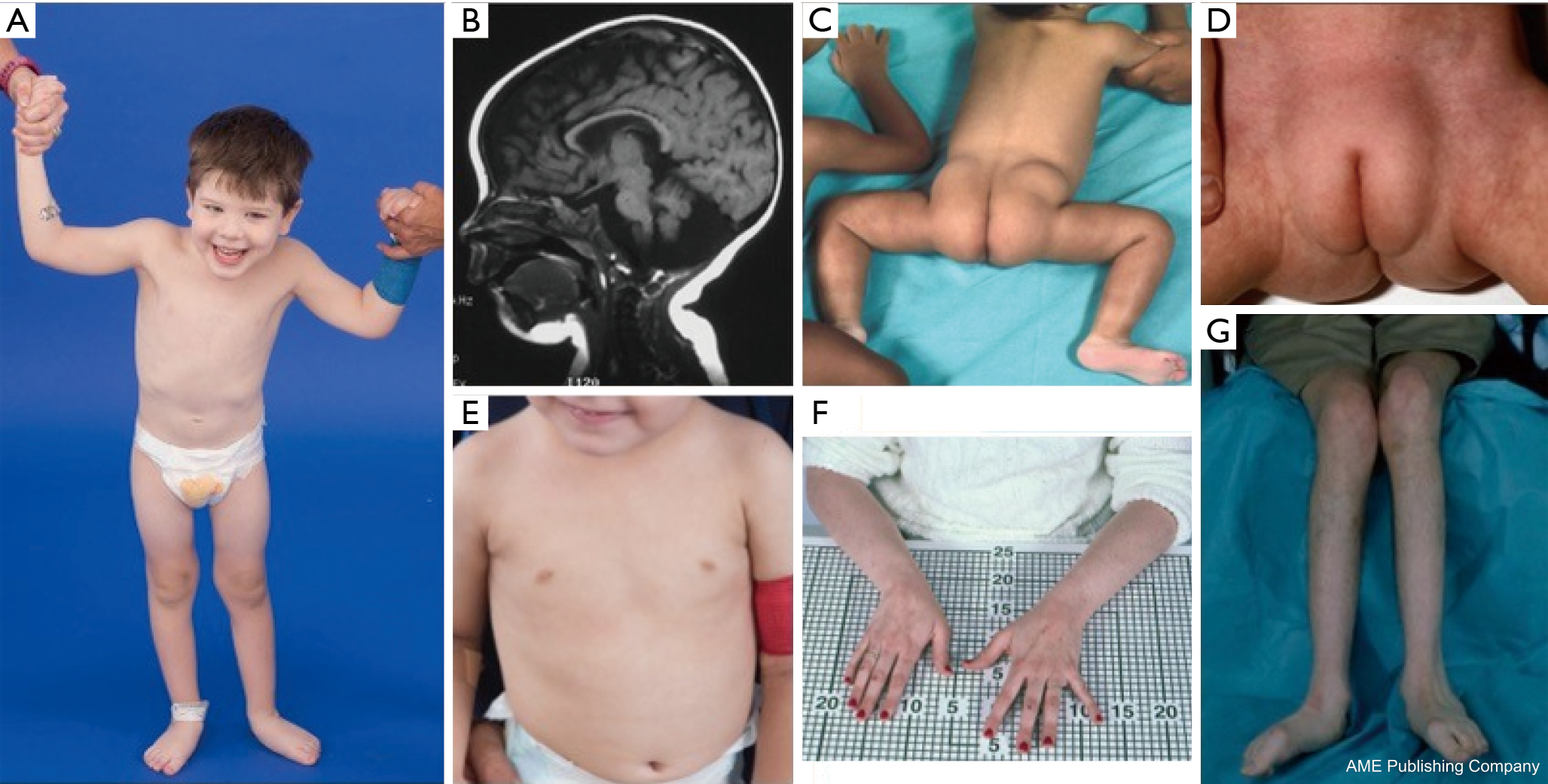
In de kindertijd kunnen getroffen personen retinitis pigmentosa, beroerte-achtige episoden en toevallen, spraak- en motorische vertragingen, en perifere neuropathie ontwikkelen. Patiënten hebben vaak een groeiachterstand als gevolg van voedings- en maagdarmafwijkingen en een algemene ontwikkelingsachterstand. Verhoogde levertransaminasen zonder klinische gevolgen kunnen worden waargenomen, die normaliter normaliseren tegen de leeftijd van 5 jaar met af en toe schommelingen met ziekte (21,24). Leverbiopsies zijn zelden geïndiceerd bij CDG tenzij leverfibrose wordt vermoed (1). Klinische hypothyreoïdie is zeldzaam, maar patiënten met CDG moeten hun schildklierhormonen en vrij T4 laten meten, waarbij een laag schildklierbindend globuline (TBG) en voorbijgaande verhogingen van het schildklierstimulerend hormoon (TSH) kunnen worden aangetoond (37). Lever- en galgangmisvormingen zijn niet gemeld bij PMM2-CDG patiënten.
Volwassenen met PMM2-CDG kunnen tot hun 7e of 8e decennium leven met een stabiele cognitieve achterstand, perifere neuropathie, en progressieve thoracale en spinale kyphoscoliose met osteopenie of osteoporose (34). Cerebellaire ataxie is een steeds meer herkend symptoom samen met multisystemische betrokkenheid (38-40). Endocriene afwijkingen waaronder hyperprolactinemie, groeihormoonafgifte met hyperglykemie, insulineresistentie, en hyperinsulinemische hypoglykemie (41,42). Bij getroffen vrouwen kan hypogonadotroop hypogonadisme leiden tot afwezige secundaire seksuele ontwikkeling of afwezige eierstokken (41,43,44). Patiënten kunnen een verhoogd risico op trombose hebben door verlaagde serum stollingsfactoren waaronder factoren IV, IX, en XI, antitrombine III, proteïne C, en proteïne S (29).
MPI-CDG (CDG-Ib, mannosefosfaat isomerase deficiëntie)
MPI-CDG is uniek omdat getroffen patiënten weinig tot geen neurologische betrokkenheid hebben en sommige manifestaties van de ziekte behandelbaar zijn met orale mannose (2). De symptomen zijn voornamelijk hepatisch-intestinaal zonder dysmorfe kenmerken of cognitieve achterstand. Patiënten presenteren zich meestal met recidiverend braken, significante hypoglykemie, falen om te gedijen, potentieel levensbedreigende eiwitverspillende enteropathie, leverfibrotische veranderingen, en galwegverwijding (45-51). Patiënten lopen een verhoogd risico op trombotische voorvallen door lage serumconcentraties van proteïne C en S, en anti-trombine III.
ALG6-CDG (glucosyltransferase 1 deficiëntie)
ALG6-CDG is het tweede meest voorkomende N-glycosyleringsdefect, gekenmerkt door een vergelijkbaar maar milder fenotype dan PMM2-CDG. Patiënten met ALG6-CDG hebben een gebrek aan gedijen, ontwikkelingsachterstand, hypotonie, toevallen, strabismus, ataxie, coagulopathie, en gezichtsdysmorfismen (d.w.z. laag aangezette oren, hypertelorisme, en macroglossie). Net als bij MPI-CDG kunnen zij ook eiwit-verliezende enteropathie hebben. Bovendien kunnen getroffen patiënten skeletafwijkingen hebben, waaronder brachydactylie en vingermalformaties en scoliose. Aangetaste patiënten hebben doorgaans geen retinitis pigmentosa of cerebellaire hypoplasie (52).
O-linked glycosylation defects and combined N- and O-linked glycosylation defects
Door de substantiële aanwezigheid van O-glycanen in mucine-bevattende eiwitten waaronder glycosaminoglycanen (GAG’s) en epitheliale oppervlakken (53), leiden stoornissen in de GAG-synthese typisch tot skeletdysplasieën of bindweefselaandoeningen. Getroffen patiënten kunnen naast neurologische symptomen ook afwijkingen aan het bewegingsapparaat, de huid en de gewrichten vertonen (bijv. gewrichtslaxiteit, multipele exostosen, chondro/osteosarcoom) (54-56). Bijvoorbeeld, N-acetylgalactosaminyltransferase 3 (GALNT3) O-glycosyliseert het fosfaathormoon, FGF23, waardoor proteolytische splitsing wordt voorkomen en de intacte secretie mogelijk wordt. GALNT3-deficiëntie leidt tot familiaire tumorale calcinose, gekenmerkt door hyperfosfatemie en ectopische calcificaties (57,58).
Lipideglycosylerings- en GPI-ankerbiosynthesedefecten
Glycosfingolipiden en hun gesialyleerde derivaten, gangliosiden, worden voornamelijk door neuronen tot expressie gebracht. Defecten in de afbraak van gangliosiden leiden tot ophoping en de goed gekarakteriseerde lysosomale opslagziekten. Daarentegen zijn defecten in de biosynthese van gangliosiden, zoals ST3GAL5-CDG en B4GALNT1-CDG, uiterst zeldzaam en leiden tot ernstige neurodegeneratieve aandoeningen. Patiënten kunnen spastische paraplegie, ernstige intellectuele achterstand, epilepsie en niet-neurologische symptomen vertonen, waaronder skeletdysplasie, dysmorfe kenmerken en abnormale huidpigmentatie (59,60).
Mutaties in veel genen binnen de biosynthese van GPI-ankers veroorzaken een verscheidenheid aan meervoudige aangeboren afwijkingen, intellectuele beperkingen en epilepsie. Het best gekarakteriseerde GPI biosynthese defect, X-gebonden PIGA deficiëntie, presenteert zich met infantiele spasmen met hypsarrhythmia, hypotonie, meerdere hersenafwijkingen, en gezichtsdysmorfismen. Patiënten kunnen ook wisselende huid-, lever-, hart- en nierziekten hebben (61-69). Sommige mutaties binnen PIGA veroorzaken de fenotypisch verschillende ziekte paroxysmale nachtelijke hemoglobinurie (PNH), een verworven aandoening van beenmergfalen (70,71).
Diagnose
Wanneer een CDG klinisch wordt vermoed, is de eerste stap het bestellen van biochemische CDG-tests in plasma of serum, waaronder CDT- en N-glycantests. Serum CDT- en N-glycaananalyse kunnen alleen N-glycosyleringsdefecten opsporen en zijn daarom niet nuttig om geïsoleerde O-glycosylerings- of GPI-verankeringsdefecten te onderscheiden. Transferrine isovorm analyse werd oorspronkelijk verkregen door iso-elektrische focussering van transferrine, aangezien falen van N-glycan synthese een gedeeltelijk tekort aan siaalzuur veroorzaakt, waardoor de lading op serumtransferrine verandert en vervolgens de kathodale migratie op een elektroforetisch veld. Op massaspectrometrie gebaseerde analyse van transferrine en N-glycan hebben nu echter isoelectrische focussering grotendeels vervangen door het identificeren van specifieke veranderingen in oligosacchariden door massa en lading (72).
N-gekoppelde proteïneglycosyleringsdefecten
Serum transferrine CDT resultaten worden gerapporteerd als de verhouding van mono-oligosaccharide/di-oligosaccharide transferrine, a-oligosaccharide/di-oligosaccharide-transferrine, tri-sialo/di-oligosaccharide-transferrine, apolipoproteïne CIII-1/apolipoproteïne CIII-2, en apolipoproteïne CIII-0/apolipoproteïne CIII-2-verhouding. Deze kwantitatieve resultaten zullen ook gepaard gaan met een interpretatie van het patroon van de bevindingen.
Een type I patroon transferrine CDT wordt gekenmerkt door verhoogde di- en asialotransferrine banden, en wijst op defecten in de N-glycansynthese in het cytosol of endoplasmatisch reticulum. Een type II patroon wordt gekenmerkt door verhoogde di- en asialotransferrine banden, en tri- en/of monosialotransferrine banden, en wijst op defecten in N-glycan verwerking in het Golgi-apparaat (73).
Als een type I serum transferrine CDT patroon wordt gedetecteerd, moet PMM2-deficiëntie of MPI-deficiëntie op de voorgrond van differentiëlen staan, omdat PMM2-CDG de meest voorkomende CDG is en MPI-CDG behandelbaar en potentieel fataal is als het niet behandeld wordt. Om de diagnoses van elkaar te onderscheiden moeten N-glycaanprofielen, moleculaire sequencing of enzymatische tests worden uitgevoerd. De diagnose PMM2-CDG of MPI-CDG wordt bevestigd door moleculaire tests die biallelische pathogene varianten in PMM2 of MPI aantonen, gevolgd door PMM- of MPI-enzymactiviteit in leukocyten of fibroblasten indien de pathogeniteit van de genetische varianten niet zeker is. N-glycaananalyse of moleculaire analyse zou de meerderheid van ALG-CDG onderscheiden van PMM2- of MPI-CDG (15).
Een type II serumtransferrine CDT-patroon wijst op Golgi-defecten zoals N-acetylglucosaminyltransferase (GnT) II-deficiëntie (CDG type IIA, MGAT2-CDG). Apolipoproteïne CIII (Apo-CIII) isovorm analyse is een aanvullende test voor een type II CDT profiel, omdat het mucine type O-glycosyleringsdefecten in het Golgi-apparaat meet. CDT of Apo-CIII hebben een beperkte gevoeligheid voor het opsporen van type II CDG. Daarom moeten N-glycaan en O-glycaan profilering en moleculair panel of exoomsequencing worden uitgevoerd wanneer deze klinische tests beschikbaar zijn. Transferrine-glycosylatiepatronen kunnen sporadisch normaliseren; daarom kunnen herhaalde tests aangewezen zijn bij patiënten met een hoge verdenkingsindex. Valse positieven kunnen worden verkregen bij patiënten met een acute crisis van erfelijke fructose-intolerantie, galactosemie, acute leverziekte, en sommige bacteriële infecties. Geen van de biochemische CDG-tests kan alle CDG’s opsporen, zodat zelfs in aanwezigheid van normale screeningresultaten moleculaire genpaneltests of exoomsequencing kunnen worden uitgevoerd bij sterke klinische verdenking. Omgekeerd zijn biochemische en functionele bevestiging van moleculair genetische bevindingen ook essentieel, aangezien de meerderheid van de patiënten met CDG ten minste één milde en vaak nieuwe missense mutatie draagt.
O-linked glycosylatiedefecten en gecombineerde N- en O-linked glycosylatiedefecten
Diagnose berust op moleculaire sequencing, aangezien transferrine isovorm analyse geïsoleerde O-glycosylatiedefecten niet zou detecteren. Gecombineerde N- en O-gekoppelde glycosyleringsdefecten kunnen worden opgespoord door CDT, ApoCIII-analyse, en plasma N-glycan en O-glycan analyse.
Lipideglycosylering en GPI-anker biosynthese defecten
Flowcytometrie van bloedgranulocyten meet de celoppervlak expressie van GPI-geankerde eiwitten zoals CD16 en CD24. Flowcytometrie-analyse van witte bloedcellen of rode bloedcellen voor bepaalde GPI-geankerde celoppervlak-eiwitten zijn klinisch beschikbaar als een test voor PNH als gevolg van verworven mutaties in het PIGA-gen. De PNH-test kan afwijkingen in andere GPI-anker-deficiënties aan het licht brengen, maar de diagnose berust meestal op moleculaire analyse.
Moleculaire analyse
Het hoogste diagnostische rendement voor CDG is een op de volgende generatie gebaseerd gensequencingpanel of klinische exoomsequencing (CES). Genetische sequencing controleert de nucleotidenvolgorde, of “letter”-spelling, van genen om te bepalen of er een verandering is die de functie van het gen beïnvloedt. Het menselijk genoom bestaat uit 3 miljoen nucleotiden, maar slechts 1-2% daarvan, de zogenaamde exonen, worden vertaald in een functioneel eiwitproduct. Het resterende niet-coderende DNA tussen de exonen, dat niet vertaald wordt, wordt intron genoemd (74). CES onderzoekt bijna alle bekende exonen van de ongeveer 20.000 genen in het menselijk genoom, die een minderheid vormen van het genetisch materiaal in chromosomen, maar die het meest waarschijnlijk ziekteveroorzakende (pathogene) varianten bevatten. CES kan ook sequencing van mitochondriaal DNA (mtDNA) omvatten, waarmee klein, extranucleair, circulair DNA in de mitochondriën wordt onderzocht, dat uitsluitend maternaal wordt overgeërfd.
De mogelijke resultaten voor CES omvatten positieve, negatieve en varianten van onbekende significantie. Een positief resultaat betekent dat bekende ziekteveroorzakende (d.w.z. pathogene) varianten zijn geïdentificeerd, waarna de diagnose, de natuurlijke geschiedenis, de prognose, het recidiverisico en de behandelingsopties kunnen worden besproken. Een negatief resultaat betekent dat er geen detecteerbare pathogene varianten werden geïdentificeerd. Varianten van onbekende betekenis (ook wel VUS genoemd) betekent dat er weliswaar genetische veranderingen zijn geïdentificeerd, maar dat er niet genoeg informatie is over de specifieke genetische verandering om definitief te weten of deze ziekteverwekkend is. Variaties in het DNA van elk individu zijn te verwachten, dus het gelijktijdig testen van ouderlijke monsters ter vergelijking kan helpen bij de laboratorium- en klinische interpretatie van de resultaten. Het diagnostische rendement van CES wordt geschat op 30-35% en neemt in de loop van de tijd toe naarmate de ontdekking van genen en de kennis over het menselijk genoom voortschrijden (75-77). CES wordt in toenemende mate besteld als de eerstelijns brede genetische test van keuze gezien de snelle doorlooptijd en lage relatieve kosten voor de hoeveelheid geanalyseerde genetische informatie. De beperkingen van CES omvatten het gebrek aan 100% gevoeligheid, het onvermogen om bepaalde soorten genetische veranderingen op te sporen (bijv. deleties, duplicaties, trinucleotide herhalingen, diepe intronic mutaties of methyleringsdefecten), en het feit dat een diagnose mogelijk geen aanvullende informatie over de ziekte oplevert of het management verandert.
In de rapportage van CES kunnen incidenteel ontdekte pathogene varianten in genen die geassocieerd zijn met bekende genetische aandoeningen, als secundaire bevindingen worden gerapporteerd (78). Deze lijst van aanbevolen ziekten is samengesteld door het American College of Medical Genetics (ACMG). De Genetic Information Nondiscrimination Act (GINA) is een belangrijke overweging bij de beslissing om al dan niet te kiezen voor het leren van incidentele bevindingen (79). GINA beschermt individuen tegen het misbruik van genetische informatie in ziektekostenverzekering en werkgelegenheid, maar niet levensverzekeringen. GINA beschermt de volgende genetische informatie: medische familiegeschiedenis, dragerschapstests, prenatale genetische tests, vatbaarheids- en voorspellende tests, en analyse van tumoren of andere beoordelingen van genen, mutaties, of chromosomale veranderingen.
Behandeling
Het beheer van CDG hangt grotendeels af van de specifieke symptomen van het individu. Terugkerende symptomen bij patiënten met CDG zijn onder meer het niet goed gedijen, algehele ontwikkelingsachterstand, braken, beroerte-achtige episoden en skeletafwijkingen. Klinische of subklinische coagulopathie, endocrinopathie, hepatopathie, en hartafwijkingen worden ook vaak gezien. Baselinelaboratoriumonderzoek om de omvang van de ziekte vast te stellen en routinecontrole worden aanbevolen, vooral voor PMM2-CDG. Deze omvatten leverfunctietests, serumalbumine, schildklierfunctietests inclusief vrij T4, proteïne C, proteïne S, antitrombine III, factor IX, urineonderzoek, en serum gonadotropines en groeihormoon.
Aanbevolen beeldvorming omvat echocardiogram, nier-echografie, botleeftijd, oftalmologisch onderzoek voor evaluatie van lens, retina, oculaire mobiliteit, en intraoculaire druk. Tenzij anders aangegeven, worden routinevaccinaties aanbevolen voor volwassenen en kinderen die aan CDG lijden. Antilichaamtiters moeten worden verkregen na vaccinatie omdat patiënten een suboptimale immunogene respons kunnen hebben. Profylactische suppletie van stollingsfactoren voorafgaand aan chirurgische ingrepen kan noodzakelijk zijn als er bij aanvang tekorten bestaan.
Evaluatie van de klinische genetica moet worden ondernomen om de erfelijke aspecten van CDG te bespreken, evenals het opzetten van een medisch centrum voor deze complexe patiënten. Het medisch centrum is meestal de dienst biochemische genetica, hoewel afdelingen genetica, neurologie of neurologie ook in deze hoedanigheid hebben gefungeerd als een specifieke dienst biochemische genetica niet beschikbaar is. Doorverwijzing van specialisten voor gastro-enterologie, hematologie, endocrinologie, voedingsondersteuning, spraak-, ergo-, fysio- en voedingstherapieën, orthopedie en revalidatiegeneeskunde is vaak noodzakelijk.
Gerichte therapieën en prognose
De behandeling van de meeste CDG-types is grotendeels ondersteunend, met enkele uitzonderingen. MPI-CDG is de meest effectieve behandeling van alle CDG. Oraal toegediende mannose wordt door intracellulaire hexokinases omgezet in mannose-6-fosfaat, waardoor de enzymatische blokkade wordt omzeild en het ontbrekende substraat wordt geproduceerd. Mannosesuppletie begint gewoonlijk met 1 g/kg lichaamsgewicht per dag, verdeeld over 4-6 doses per dag. Terwijl de potentieel levensbedreigende eiwitverspillende enteropathie bijzonder goed reageert op mannosebehandeling, kan de leverziekte bij MPI-CDG blijven voortschrijden. Klinische symptomen verbeteren snel en transferrine CDT normaliseren in de loop van maanden, hoewel de leverziekte kan blijven voortschrijden met de behandeling (45,80,81).
Voorzichtigheid is geboden bij het toedienen van mannose tijdens de zwangerschap, aangezien toediening van mannose bij zwangere hypomorfe fosfomannose-isomerase muismodellen resulteerde in embryonale letaliteit en blindheid bij hun pups (82). Bovendien is intraveneuze mannose in verband gebracht met verminderd bewustzijn en toevallen, die verdwenen na toediening van glucose (83).
De behandeling voor PMM2-CDG is grotendeels ondersteunend en gebaseerd op symptomatologie. Momenteel worden echter klinische proeven met een vervangtherapie voor mannose-1-fosfaat substraat ontwikkeld.
Voor andere CDG zijn verschillende orale enkelvoudige suikers onderzocht met het doel de hypoglycosylering theoretisch te verbeteren. Fucose is geprobeerd voor SLC35C1-CDG en galactose voor PGM1-CDG en SLC35A2-CDG met gemengde resultaten (84). Van D-galactose van 1,0-2,5 g/kg/dag (max 50 gram) is aangetoond dat het de hypoglykemie, coagulopathie en endocrinopathie bij PGM1-CDG verbetert (85,86). Van galactose is ook aangetoond dat het de endocrinopathie en coagulopathie verbetert bij TMEM165-CDG (87) en SLC39A8-CDG. Aanzienlijke klinische verbetering werd ook gemeld bij SLC39A8-CDG-patiënten die 15-20 mg/kg/dag MnSO4 kregen (88). Er zijn klinische studies aan de gang om de bruikbaarheid van N-acetylmannosamine (ManNAc) bij GNE-CDG (89) te onderzoeken, en er zijn verschillende preklinische studies aan de gang voor andere CDG (90).
Ondanks medische vooruitgang bestaat er een aanzienlijke mortaliteit voor kinderen met CDG binnen het eerste levensjaar door multi-orgaanfalen of ernstige infectie (91). Zuigelingen met CDG kunnen zich presenteren met fulminante multi-orgaanziekte, hardnekkige toevallen, of ernstige hypoalbuminemie die overgaat in anasarca. Sommige patiënten reageren op agressieve diurese en albuminevervanging, terwijl anderen de behandeling afwijzen. Van natriumbutyraat is aangetoond dat het de controle over aanvallen verbetert bij CAD-CDG en PIGM-CDG (92). Van een ketogeen dieet is ook aangetoond dat het de aanvalsfrequentie vermindert in sommige gevallen van PIGA-CDG (93). Tijdens beroerte-achtige episoden kunnen intraveneuze hydratatie en het handhaven van normale bloedglucose nuttig zijn terwijl onderliggende vasculaire trombotische of bloedende etiologie wordt uitgesloten.
Met de komst van genome-editing technieken en een beter begrip van het mechanisme van ziekten die onder de diagnostische paraplu van CDG vallen, blijft de toekomst van gerichte therapeutische ontwikkeling veelbelovend.
Acknowledgements
We willen Lynne Wolfe, ARNP en Donna Krasnewich, MD, PhD bedanken voor het beschikbaar stellen van klinische foto’s die zijn verkregen als onderdeel van de Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). We willen ook Jenny Thies, MS, LGC bedanken voor haar expertise in genetische counseling.
Financiering: IJ Chang wordt ondersteund door de National Institutes of Health T32GM007454.
Footnote
Conflicts of Interest: De auteurs hebben geen belangenconflicten aan te geven.
Informed Consent: Schriftelijke geïnformeerde toestemming werd verkregen van de patiënten voor publicatie van dit manuscript en eventuele bijbehorende afbeeldingen.
- Jaeken J, Matthijs G. Aangeboren aandoeningen van de glycosylering. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenitale aandoeningen van de glycosylatie: een snel groeiende ziekte familie. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutaties in PMM2 die congenitale stoornissen van de glycosylatie, type Ia (CDG-Ia) veroorzaken. Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Gebrek aan Hardy-Weinberg-evenwicht voor de meest voorkomende PMM2-mutatie in CDG-Ia (congenitale stoornissen van de glycosylatie type Ia). Europees Tijdschrift voor Menselijke Genetica 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG-nomenclatuur: tijd voor verandering! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Wat is nieuw in CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Aangeboren aandoeningen van de glycosylering. Deel I. Defecten van eiwit N-glycosylatie. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellulaire functies van N-gekoppelde glycanen. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Klinische, laboratorium- en moleculaire bevindingen en follow-upgegevens op lange termijn bij 96 Franse patiënten met PMM2-CDG (fosfomannomutase 2-congenitale stoornis van de glycosylering) en overzicht van de literatuur. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG in Zuid-Afrika: Genotype-Fenotype Beschrijving van Vijf Nieuwe Patiënten. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Different glycosyltransferases are differentially processed for secretion, dimerization, and autoglycosylation. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In: Mora-Montes H. Editor. Glycanen: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenitale stoornissen van de glycosylatie: andere oorzaken van ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Huidmanifestaties bij CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF-systeem bij kinderen met congenitale stoornissen van de glycosylatie. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertal growth in congenital disorder of glycosylation type Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Klinische en biochemische kenmerken van congenitale stoornis van de glycosylatie type Ic, het eerste erkende endoplasmatisch reticulum defect in N-glycan synthese. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–een recent ontdekte erfelijke stofwisselingsziekte. Meerdere orgaan manifestaties, incidentie 1/80.000, moeilijk te behandelen. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Pericardiale en abdominale vochtophoping bij congenitale stoornis van de glycosylatie type Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Klinische kenmerken bij volwassenen met aangeboren stoornissen van de glycosylatie type Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Carbohydrate-deficient glycoprotein syndrome. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modification of the coat protein of the potyvirus Plum pox virus enhances viral infection. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Scandinavische CDG-Ia patiënten: genotype/fenotype correlatie en geografische oorsprong van stichtermutaties. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 Franse volwassen patiënten met PMM2-congenitale stoornis van de glycosylering: uitkomst van het klassieke pediatrische fenotype en depictie van een late-onset fenotype. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinale kenmerken van de congenitale stoornis van de glycosylatie PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenitale stoornis van de glycosylatie type Ia (CDG-Ia): fenotypisch spectrum van het R141H/F119L genotype. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Schildklierfunctie bij PMM2-CDG: diagnostische benadering en voorgesteld management. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mentale ontwikkeling bij een congenitale stoornis van de glycosylatie (CDG) type Ia patiënt met multisystemische betrokkenheid (intermediair fenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenitale stoornis van de glycosylatie type 1a: drie broers en zussen met een mild neurologisch fenotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. New disorders in carbohydrate metabolism: congenital disorders of glycosylation and their impact on the endocrine system. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenitale stoornis van de glycosylatie type Ia: heterogeniteit in de klinische presentatie van multiviscerale uitval tot hyperinsulinemische hypoglykemie als leidende symptomen bij drie zuigelingen met fosfomannomutase-deficiëntie. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadal function and glycoprotein hormones in the carbohydrate-deficient glycoprotein (CDG) syndrome. Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Het klinisch spectrum van fosfomannose isomerase deficiëntie, met een evaluatie van mannose behandeling voor CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Cardiale complicaties van congenitale stoornissen van de glycosylatie (CDG): een systematisch overzicht van de literatuur. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Fosfomannose isomerase deficiëntie en mannose therapie. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: A new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Klinische bruikbaarheid genenkaart voor: ALG6 defecte congenitale stoornis van de glycosylatie. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: Van productie tot secretie. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparan sulfate in skelet development, growth, and pathology: the case of hereditary multiple exostoses. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Diverse niet-inflammatoire musculoskeletale aandoeningen. Hyperfosfatemische familiaire tumorcalcinose (FGF23, GALNT3 en αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Ablatie van het Galnt3 gen leidt tot lage circulerende intacte fibroblast groeifactor 23 (Fgf23) concentraties en hyperfosfatemie ondanks verhoogde Fgf23 expressie. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Een mutatie in een ganglioside biosynthetisch enzym, ST3GAL5, resulteert in zout & peper syndroom, een neurocutane aandoening met veranderde glycolipide en glycoproteïne glycosylatie. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutaties in B4GALNT1 (GM2 synthase) liggen ten grondslag aan een nieuwe stoornis in de biosynthese van gangliosiden. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA-mutaties veroorzaken vroeg ontstane epileptische encefalopathieën en onderscheidende kenmerken. Neurology. Neurologie 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome wordt veroorzaakt door een mutatie in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorfe promotormutatie in PIGM veroorzaakt erfelijke glycosylphosphatidylinositol-deficiëntie. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorfe mutaties in PGAP2, coderend voor een GPI-anchor-remodeling protein, veroorzaken autosomaal-recessieve verstandelijke handicap. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. Het fenotype van een kiembaanmutatie in PIGA: het gen dat somatisch gemuteerd is in paroxysmale nachtelijke hemoglobinurie. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutaties in PIGO, een lid van de GPI-anchor-synthesis pathway, veroorzaken hyperfosfatasie met mentale retardatie. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutaties in het glycosylfosfatidylinositol-gen PIGL veroorzaken CHIME-syndroom. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. Het genotypische en fenotypische spectrum van PIGA-deficiëntie. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmale nachtelijke hemoglobinurie (PNH) wordt veroorzaakt door somatische mutaties in het PIG-A gen. EMBO J 1994;13:110-7.
- Brodsky RA. Paroxysmale nachtelijke hemoglobinurie. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. De impact van massaspectrometrie in de diagnose van congenitale stoornissen van de glycosylatie. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomaal recessieve gedilateerde cardiomyopathie als gevolg van DOLK-mutaties resulteert uit abnormale dystroglycaan O-mannosylering. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Stromingen in de hedendaagse ethiek. GINA, de ADA, en genetische discriminatie bij tewerkstelling. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Recurrent trombo-embolie bij een kind met een congenitale stoornis van de glycosylatie (CDG) type Ib en behandeling met mannose. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Ontwikkeling van leverziekte ondanks behandeling met mannose bij twee patiënten met CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplementen induceren embryonale letaliteit en blindheid in phosphomannose isomerase hypomorphic muizen. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Aanvallen en stupor tijdens intraveneuze mannosetherapie bij een patiënt met CDG-syndroom type 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Fucosesuppletie bij leukocyt-adhesiedeficiëntie type II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Inherited glycosylphosphatidyl inositol deficiency: a treatable CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Orale D-galactose suppletie bij PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Galactose Suppletie bij Patiënten met TMEM165-CDG Rescues the Glycosylation Defects. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8-deficiëntie: biochemische correctie en belangrijke klinische verbetering door mangaantherapie. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Human glycosylation disorders. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Gerichte therapie voor erfelijke GPI-deficiëntie. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev 2016;38:848-51.