- Panoramica
- Epidemiologia
- Classificazione biochimica e nomenclatura
- Genetica
- Patofisiologia
- Difetti di glicosilazione delle proteine legate all’N
- difetti di glicosilazione O-linked e difetti di glicosilazione combinati N- e O-linked
- Difetti di glicosilazione dei lipidi e biosintesi dell’ancora GPI
- Segnalazioni cliniche
- N-collegato difetti di glicosilazione delle proteine
- PMM2-CDG (CDG-Ia, deficit di PMM2)
- MPI-CDG (CDG-Ib, deficit di mannosefosfato isomerasi)
- ALG6-CDG (deficit di glucosiltransferasi 1)
- Difetti di glicosilazione O-linked e difetti combinati di glicosilazione N- e O-linked
- Difetti di glicosilazione dei lipidi e biosintesi dell’ancora GPI
- Diagnosi
- difetti di glicosilazione delle proteine N-collegate
- Difetti di glicosilazione O-linked e difetti combinati di glicosilazione N- e O-linked
- Difetti di glicosilazione dei lipidi e biosintesi di ancore GPI
- Analisi molecolare
- Gestione
- Terapie mirate e prognosi
- Riconoscimenti
- Footnote
Panoramica
La glicosilazione è il processo di aggiunta di residui di zucchero a proteine e lipidi in diversi percorsi cellulari. I disordini congeniti della glicosilazione (CDG) sono un gruppo geneticamente e clinicamente eterogeneo di oltre un centinaio di malattie causate da difetti in varie fasi della sintesi dei glicani o delle vie di modifica. La maggior parte di queste malattie monogeniche sono autosomica recessiva in eredità, ma autosomica dominante e X-linked forme sono state descritte anche.
CDG tipicamente presentano con manifestazioni multi-sistema, più comunemente ritardo nello sviluppo, mancata crescita, ipotonia, anomalie neurologiche, epatopatia e coagulopatia. Gli individui affetti possono anche presentare malattie oculari, cutanee e cardiache, così come dismorfismi facciali. Anche se i cambiamenti neurologici e i ritardi cognitivi sono visti nella maggior parte degli individui affetti, ci sono alcuni casi e anche tipi che non hanno manifestazioni neurologiche. Data l’ampia eziologia clinica e genetica della CDG, la diagnosi clinica si basa su un alto indice di sospetto nella malattia multisistemica.
L’analisi della transferrina carente di carboidrati nel siero (CDT) è il test di screening di prima linea nei pazienti con sospetta CDG, ma è limitata nella rilevazione dei difetti di N-glicosilazione con carenze di acido sialico. I test successivi includono l’analisi dei glicani legati al dolicol e i test genetici. La diagnosi precoce di questo gruppo di malattie in crescita esponenziale è importante, poiché alcune CDG sono curabili. Il trattamento per i difetti di glicosilazione è principalmente di supporto, sebbene siano disponibili terapie mirate per MPI-CDG, SLC35C1-CDG, PIGM-CDG e PGM1-CDG. I dettagli su questi trattamenti sono nella sezione “Terapie mirate e prognosi” qui sotto. L’attenzione di questa revisione sarà sui tipi più comuni di CDG con fenotipi o trattamenti riconoscibili, con il pubblico target di fornitori di cure primarie.
Epidemiologia
L’incidenza e la prevalenza di tutti i tipi di CDG in aggregato non sono state ben stabilite, anche se i pazienti sono stati segnalati in tutto il mondo da quasi ogni background etnico ed entrambi i sessi sono ugualmente colpiti. La prevalenza stimata nelle popolazioni europee e afroamericane è di 1/10.000 sulla base delle frequenze di portatori di varianti patogene note in 53 geni (1-4). La prevalenza della CDG più comunemente diagnosticata, la PMM2-CDG, varia da 1/20.000 nelle popolazioni olandesi e 1/77.000 in Estonia sulla base di rapporti isolati (5,6). Ad oggi, sono stati riportati meno di 100 casi per la maggior parte dei tipi di CDG.
Classificazione biochimica e nomenclatura
In generale, le CDG sono attualmente classificate in quattro categorie: (I) glicosilazione N-linked, (II) glicosilazione O-linked, (III) glicosilazione combinata N- e O-linked/multipla, e (IV) lipidi e difetti di biosintesi dell’ancora glicosilfosfatidilinositolo (GPI).
Il difetto di glicosilazione proteica N-linked PMM2-CDG, precedentemente noto come CDG tipo Ia, è stato il primo CDG ad essere riportato da Jaeken nel 1980 e rimane di gran lunga il più comune CDG ad oggi (7). La PMM2-CDG è stata inizialmente denominata “sindrome da glicoproteina carente di carboidrati” a causa delle anomalie multiple delle glicoproteine sieriche osservate dalla focalizzazione isoelettrica della transferrina sierica negli individui affetti. Storicamente, le CDG sono state classificate in base ai modelli di analisi delle isoforme della transferrina: i modelli di tipo I sono stati attribuiti a difetti di assemblaggio e trasferimento dei glicani legati al dolicol e localizzati nel citoplasma o nell’ER, mentre i modelli di tipo II sono stati attribuiti a difetti di elaborazione nell’apparato di Golgi. Da questo punto di diramazione, i CDG sono stati denominati in ordine alfabetico in base alla scoperta.
Con l’avvento della diagnostica molecolare diffusa, la nomenclatura CDG è stata aggiornata nel 2008 per specificare l’eziologia molecolare della malattia, riflettendo la crescita esponenziale di percorsi e disturbi che non si adattavano perfettamente alle categorie dicotomiche precedentemente stabilite. Attualmente, la nomenclatura CDG è indicata dal nome del gene interessato (non in corsivo, i nomi dei geni in www.genenames.org), seguito da -CDG (ad esempio, PMM2-CDG) (8).
Genetica
La grande maggioranza dei disturbi congeniti della glicosilazione sono ereditati in modo autosomico recessivo, con una mutazione ereditata da ogni genitore asintomatico (portatore). I test molecolari, di solito con metodi di sequenziamento di prossima generazione, sono necessari per stabilire una diagnosi genetica. Il test parentale per la variante conosciuta può confermare l’ereditarietà rispetto alla comparsa de novo. Per l’eredità autosomica recessiva, il rischio di ricorrenza per i fratelli e ogni gravidanza di un individuo affetto è del 25% per essere affetto, il 50% per essere un portatore asintomatico, e il 25% per essere non affetto.
Una manciata di CDG hanno eredità autosomica dominante (N-linked: GANAB-CDG, PRKCSH-CDG; O-linked: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Meno sono X-linked (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). La maggior parte delle forme dominanti e alcune forme X-linked di CDG sono dovute a mutazioni de novo. Malattie e geni specifici sono descritti di seguito nella sezione “fisiopatologia”.
I dati di mutazione per tutti i geni pubblicati per la CDG sono disponibili presso lo Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Informazioni su specifiche varianti geniche sono disponibili presso il Leiden Open Variation Database con strumenti integrati di patogenicità in silico (http://www.lovd.nl/3.0/home). Sinossi cliniche per geni specifici possono essere trovate presso l’Online Mendelian Inheritance in Man (http://www.omim.org/) o in un ambito più limitato presso GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Dato il piccolo numero di pazienti affetti per la maggior parte dei sottotipi di CDG, la correlazione genotipo-fenotipo è difficile da stabilire.
Patofisiologia
Oltre 130 tipi di CDG sono stati riportati finora (9,10). Data la presenza ubiquitaria di vie di glicosilazione, le CDG sono estremamente diverse nella loro patogenesi biochimica. Numerose proteine e lipidi (cioè sfingolipidi e glicolipidi) subiscono la glicosilazione con monosaccaridi e/o oligosaccaridi, chiamati collettivamente glicani, in diversi compartimenti cellulari. Le loro posizioni subcellulari sono diverse, ma la maggior parte dei difetti si verifica all’interno dell’apparato ER o Golgi. Le caratteristiche cliniche e l’eziologia genetica delle CDG più comuni per via sono riassunte nella tabella S1.
Tra le proteine, i glicani sono descritti dal loro legame alla catena polipeptidica-N-glicani sono attaccati al gruppo amidico dell’asparagina (Asn) mentre O-glicani sono attaccati al gruppo idrossile della serina o della treonina. La sintesi degli N-glicani richiede la costruzione graduale di zuccheri legati al nucleotide nel citosol, l’assemblaggio nel reticolo endoplasmatico e l’elaborazione nell’apparato di Golgi. Al contrario, la sintesi di O-glicano richiede l’assemblaggio ma nessuna elaborazione, quindi i difetti di O-glicosilazione si verificano prevalentemente nell’apparato di Golgi.
Difetti di glicosilazione delle proteine legate all’N
La glicosilazione dell’N comporta l’attacco covalente di strutture di carboidrati al gruppo ammidico della catena laterale dei residui Asn all’interno di un sito accettante di consenso Asn-X-Ser/Thr, la traslocazione del polipeptide substrato al reticolo endoplasmatico per il rimodellamento e l’ulteriore modifica della catena N-glicano nel Golgi (11,12). Difetti in qualsiasi punto del percorso di sintesi, assemblaggio ed elaborazione possono portare alla malattia clinica.
PMM2-CDG è causato da varianti patogene nel gene della fosfomannomutasi 2 (PMM2), che porta alla carenza dell’enzima PMM2 che catalizza la conversione citosolica del mannosio-6-fosfato in mannosio-1-fosfato nella seconda fase della sintesi del mannosio con guanosina difosfato (GDP). La maggior parte dei pazienti ospita mutazioni missense eterozigoti composti patogeni (www.lovd.nl/PMM2). La variante patogena ricorrente più comune p.Arg141His si trova in circa il 40% degli individui affetti di origine europea, e p.Phe119Leu si trova anche frequentemente nel nord Europa (1). Correlazioni genotipo-fenotipo sono state riportate per PMM2-CDG (3,13,14).
MPI-CDG è un disordine autosomico recessivo causato da varianti patogene nel gene della mannosio fosfato isomerasi (MPI) che portano ad un deficit di fosfomannosio isomerasi (MPI). MPI normalmente catalizza il primo passo della sintesi del PIL-mannosio (cioè la conversione del fruttosio-6-fosfato in mannosio-6-fosfato), ma il fruttosio-6-fosfato non si accumula intracellularmente poiché può anche essere metabolizzato dalla via glicolitica. Pertanto, anche se biochimicamente simile al PMM2-CDG, l’MPI-CDG non causa un coinvolgimento neurologico e multisistemico altrettanto significativo. Il CDT è anche il test di screening di scelta per la MPI-CDG, che mostra un pattern di tipo 1. La diagnosi può poi essere confermata molecolarmente o dall’attività MPI dei fibroblasti/leucociti.
ALG6-CDG è una malattia recessiva causata da mutazioni in ALG6, che porta all’attaccamento anormale di tre molecole di glucosio agli intermedi di mannosio legati al dolicholo e all’ipoglicosilazione a valle delle glicoproteine del siero (15).
difetti di glicosilazione O-linked e difetti di glicosilazione combinati N- e O-linked
O-glicosilazione comprende l’aggiunta graduale di catene di carboidrati ai residui di serina, treonina e idrossilisina delle proteine da parte delle glicosiltransferasi nell’apparato di Golgi (16). Diversi tipi di glicani O-linked sono stati associati a malattie umane, denominati in base al primo zucchero attaccato al residuo aminoacidico (17).
Difetti di glicosilazione dei lipidi e biosintesi dell’ancora GPI
Le ancore GPI sono glicolipidi che subiscono un assemblaggio sequenziale nel reticolo endoplasmatico e modifiche all’interno del Golgi. I disturbi della biosintesi delle ancore GPI dovuti a carenze enzimatiche sono nominati in ordine alfabetico per ordine di scoperta e non cronologicamente per fase di assemblaggio. Una volta sintetizzate, le ancore GPI risiedono sulle membrane plasmatiche e legano centinaia di proteine della superficie cellulare, svolgendo una pletora di funzioni cellulari. La maggior parte di queste malattie sono autosomiche recessive con la notevole eccezione del deficit di PIGA legato all’X.
Segnalazioni cliniche
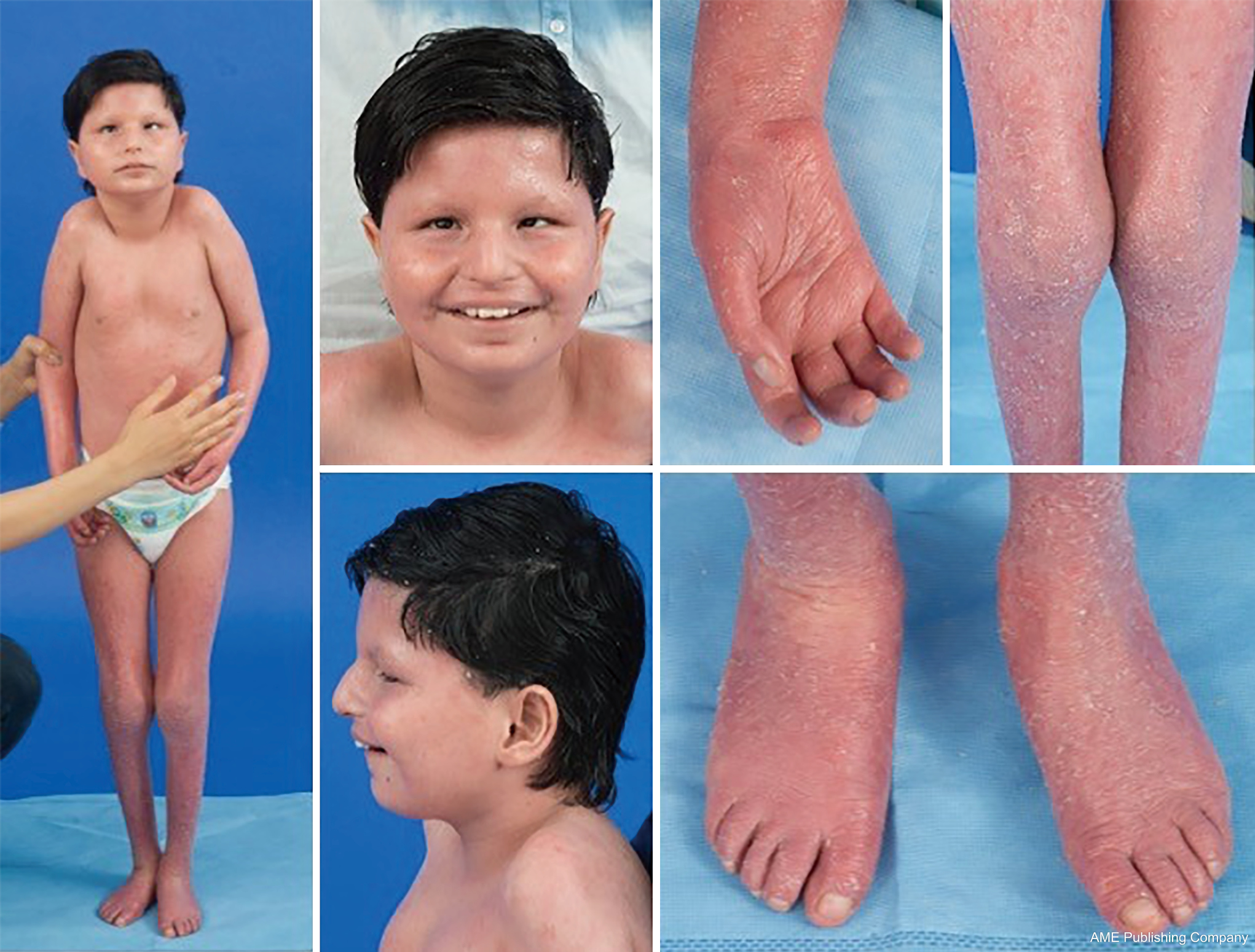
Data la presenza ubiquitaria delle vie di glicosilazione, praticamente qualsiasi sistema d’organo può essere coinvolto nella CDG, anche se la maggior parte dei casi comporta anomalie neurologiche. Alcune CDG si presentano con ittiosi, tra cui MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (Figura 1) e PIGL-CDG (18,19). Quasi tutte le CDG presentano una malattia multisistemica entro i primi anni di vita, tranne alcune che colpiscono solo un singolo sistema d’organo (es, retina in DHDDS-CDG; giunzione neuromuscolare in ALG2-CDG, ALG14-CDG, CFPT1-CDG; cervello in ST3GAL3-CDG, TUSC3-CDG; pelle o muscoli scheletrici in POGLUT1-CDG, POFUT1-CDG; cartilagine in EXT1/EXT2-CDG; fegato in TMEM199-CDG; globuli rossi in SEC23B-CDG). L’età di insorgenza e la gravità possono variare da neonatale letale a quasi asintomatico in età adulta, e qualsiasi permutazione nel mezzo. La costellazione di sintomi più comunemente riportata include ritardo nello sviluppo, mancata crescita, ipotonia, anomalie neurologiche, ipoglicemia e anomalie variabili di fegato, occhi, pelle, gastrointestinali, immunologiche, scheletriche e di coagulazione (19).
Il fenotipo completo per molti sottotipi di CDG rimane da delineare completamente a causa della rarità dei casi riportati. Pertanto, CDG dovrebbe essere considerato in qualsiasi impostazione della malattia multisistemica, soprattutto nei casi con una componente neurologica o un ritardo di sviluppo aspecifico con eziologia non chiara.
Anche se la fisiopatologia dei sintomi di moltitudine rimane da chiarire, la relazione tra alcuni percorsi di glicosilazione e sintomi clinici specifici è stata chiarita. Per esempio, l’incapacità di crescere vista in molti tipi di CDG è attribuibile all’ipoglicosilazione e alla formazione alterata di diverse glicoproteine all’interno della via di crescita dell’insulina, tra cui IGF-1, ALS e IGFBP-3 (20). Man mano che comprendiamo meglio questo gruppo di disturbi complessi, i CDG sono sempre più riconosciuti in individui con diagnosi elusive. I sistemi d’organo coinvolti nei diversi CDG sono riassunti nella Tabella S1. Discuteremo le caratteristiche cliniche delle forme più comuni e le forme con trattamenti mirati di CDG sotto.
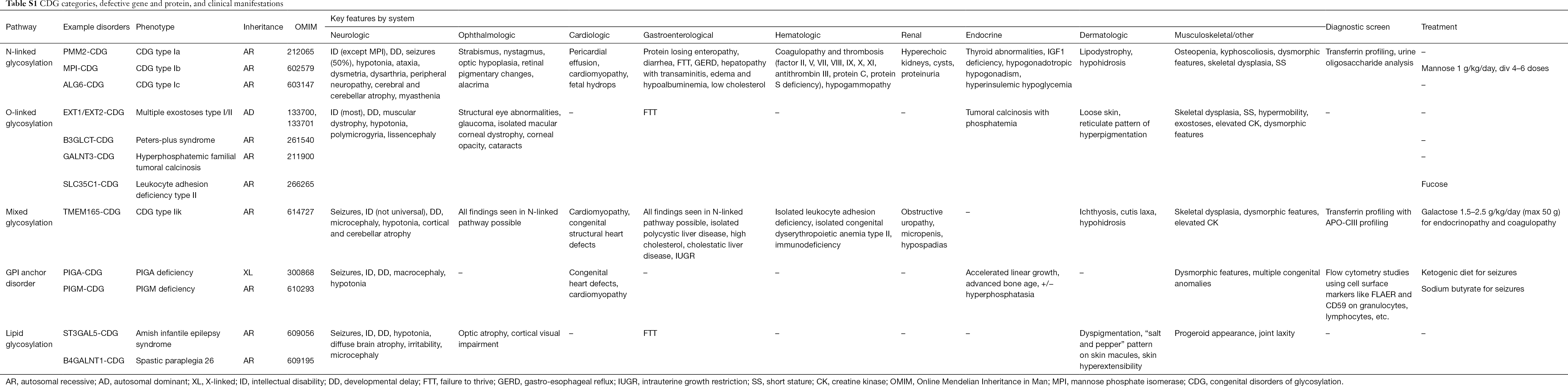
Tabella completa
N-collegato difetti di glicosilazione delle proteine
Come il più comunemente diagnosticato CDG, il fenotipo di N-linked disturbi glicosilazione è spesso annunciato come la presentazione classica. Tuttavia, lo spettro fenotipico di CDG è abbastanza diverso, e molti CDG possono non presentare i sintomi stereotipati associati a PMM2-CDG.
PMM2-CDG (CDG-Ia, deficit di PMM2)
PMM2-CDG è la CDG più comune, con oltre 700 casi riportati in tutto il mondo. È caratterizzata da una grave malattia multisistemica nell’infanzia, malattia neurologica e ritardo dello sviluppo nell’infanzia, e/o disabilità intellettuale stabile nell’età adulta (21,22).
Nell’infanzia, la PMM2-CDG si presenta con anomalie neurologiche tipicamente poco dopo la nascita, cioè strabismo e movimenti oculari anomali, ipoplasia cerebellare, ipotonia, ritardo psicomotorio, atassia, ipotonia e iporeflessia. I bambini possono anche avere malattia epatica, sindrome nefrosica e cisti renali, versamento pericardico e cardiomiopatia ipertrofica, mancata crescita e insufficienza multiorgano con conseguente morte entro il primo anno di vita fino al 20% degli individui affetti (21,23-28).
Una costellazione di caratteristiche dismorfiche è stato descritto in pazienti con PMM2-CDG (figure 2,3). Questi includono un cervelletto ipoplasico, dismorfismi facciali (cioè, grandi orecchie displasiche), capezzoli invertiti e un’anormale distribuzione del tessuto adiposo sulle natiche o sulla regione sovrapubica che può risolversi con l’età (14,21,29-32). I pazienti sono stati descritti per avere un contegno estroverso e felice. La presentazione è molto variabile, anche se lo strabismo può essere visto in più del 70% dei pazienti affetti (21,23,33-35). Capezzoli invertiti e cuscinetti di grasso anormali si vedono in circa il 25-50% dei pazienti (36).

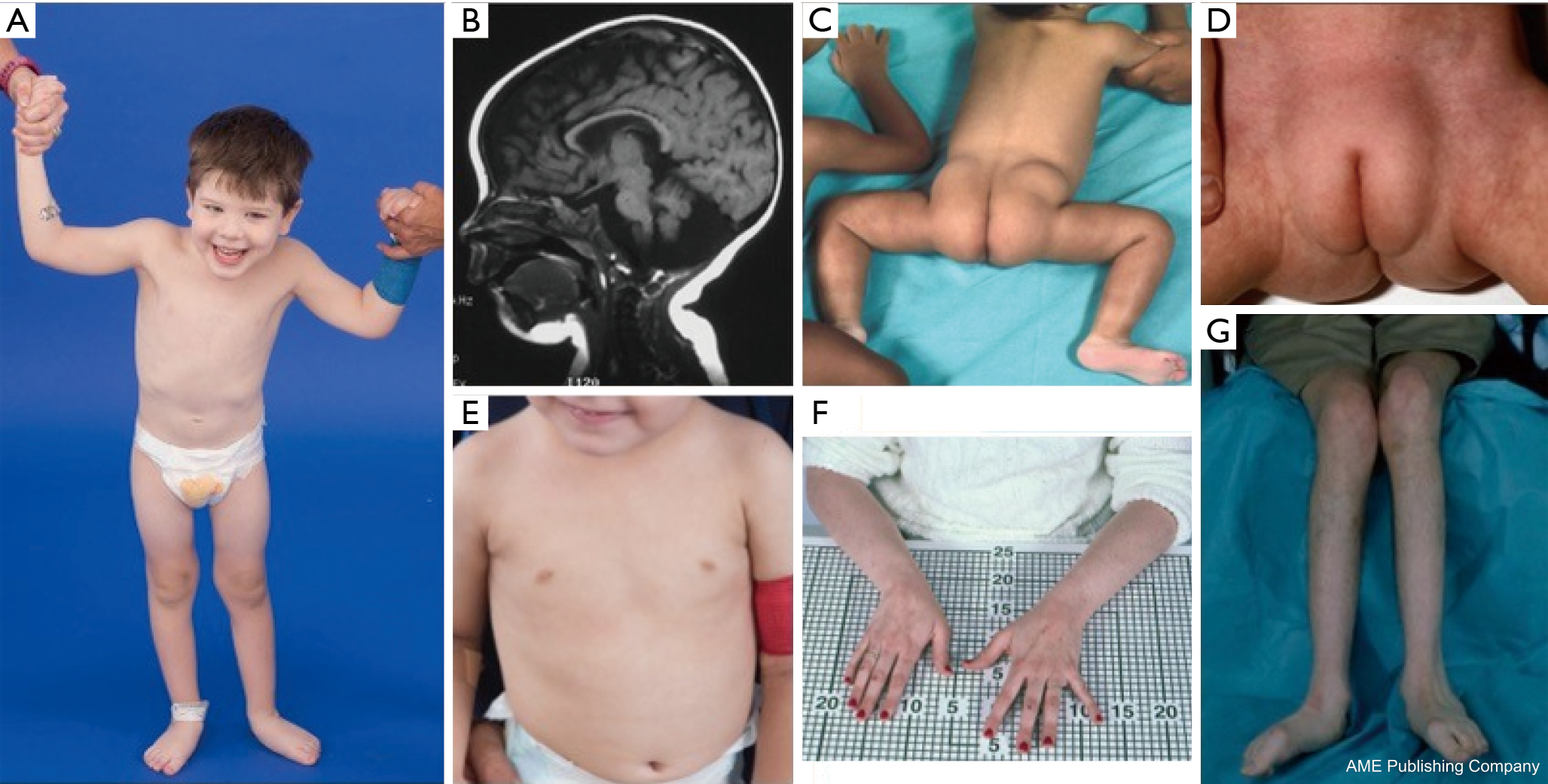
Nell’infanzia, gli individui affetti possono sviluppare retinite pigmentosa, episodi simili a ictus e convulsioni, ritardi motori e del linguaggio e neuropatia periferica. Costituzionalmente, i pazienti hanno comunemente il fallimento di prosperare a causa di anomalie di alimentazione e GI e ritardo di sviluppo globale. Possono essere osservate transaminasi epatiche elevate senza conseguenze cliniche, che in genere si normalizzano entro i 5 anni di età con occasionali fluttuazioni con la malattia (21,24). Le biopsie epatiche sono raramente indicate nella CDG a meno che non si sospetti una fibrosi epatica (1). L’ipotiroidismo clinico è raro, ma i pazienti con CDG dovrebbero farsi misurare gli ormoni tiroidei e la T4 libera, che possono mostrare una bassa globulina legante la tiroide (TBG) e aumenti transitori dell’ormone stimolante la tiroide (TSH) (37). Malformazioni del fegato e delle vie biliari non sono state riportate nei pazienti con PMM2-CDG.
Gli adulti con PMM2-CDG possono vivere fino alla settima o ottava decade con ritardo cognitivo stabile, neuropatia periferica e progressiva cifoscoliosi toracica e spinale con osteopenia o osteoporosi (34). L’atassia cerebellare è un sintomo sempre più riconosciuto insieme al coinvolgimento multisistemico (38-40). Anomalie endocrine includono iperprolattinemia, rilascio di ormone della crescita con iperglicemia, insulino-resistenza e ipoglicemia iperinsulinemica (41,42). Nelle donne affette, l’ipogonadismo ipogonadotropo può portare a uno sviluppo sessuale secondario assente o a ovaie assenti (41,43,44). I pazienti possono essere ad aumentato rischio di trombosi a causa della diminuzione dei fattori di coagulazione nel siero, compresi i fattori IV, IX e XI, antitrombina III, proteina C e proteina S (29).
MPI-CDG (CDG-Ib, deficit di mannosefosfato isomerasi)
MPI-CDG è unico perché i pazienti affetti hanno poco o nessun coinvolgimento neurologico e alcune manifestazioni della malattia sono trattabili con mannosio orale (2). I sintomi sono principalmente epatico-intestinali senza caratteristiche dismorfiche o ritardi cognitivi. I pazienti presentano tipicamente vomito ricorrente, ipoglicemia significativa, mancata crescita, enteropatia da deperimento proteico potenzialmente pericolosa per la vita, cambiamenti fibrotici epatici e dilatazione dei dotti biliari (45-51). I pazienti sono ad aumentato rischio di eventi trombotici a causa delle basse concentrazioni sieriche della proteina C e S e dell’antitrombina III.
ALG6-CDG (deficit di glucosiltransferasi 1)
ALG6-CDG è il secondo difetto di N-glicosilazione più comune caratterizzato da un fenotipo simile ma più lieve rispetto al PMM2-CDG. I pazienti con ALG6-CDG hanno mancato di crescere, ritardo nello sviluppo, ipotonia, convulsioni, strabismo, atassia, coagulopatia e dismorfismi facciali (cioè, orecchie basse, ipertelorismo e macroglossia). Simile al MPI-CDG, possono anche avere un’enteropatia da perdita di proteine. Inoltre, i pazienti affetti possono avere anomalie scheletriche tra cui brachidattilia e malformazioni delle dita e scoliosi. I pazienti affetti non hanno tipicamente retinite pigmentosa o ipoplasia cerebellare (52).
Difetti di glicosilazione O-linked e difetti combinati di glicosilazione N- e O-linked
A causa della sostanziale presenza di O-glicani nelle proteine contenenti mucina, compresi i glicosaminoglicani (GAG) e le superfici epiteliali (53), i disturbi della sintesi dei GAG portano tipicamente a displasie scheletriche o malattie del tessuto connettivo. I pazienti affetti possono presentare anomalie muscoloscheletriche, cutanee e articolari (ad esempio, lassità articolare, esostosi multiple, condro/osteosarcomi) oltre a sintomi neurologici (54-56). Per esempio, la N-acetilgalattosaminiltransferasi 3 (GALNT3) O-glicosilata l’ormone fosforico FGF23, impedendo la scissione proteolitica e permettendo la sua secrezione intatta. Il deficit di GALNT3 porta alla calcinosi tumorale familiare, caratterizzata da iperfosfatemia e calcificazioni ectopiche (57,58).
Difetti di glicosilazione dei lipidi e biosintesi dell’ancora GPI
I glicosingolipidi e i loro derivati sialilati, gangliosidi, sono espressi principalmente dai neuroni. Difetti nella scomposizione dei gangliosidi portano all’accumulo e alle ben caratterizzate malattie da accumulo lisosomiale. All’estremo opposto, i difetti nella biosintesi dei gangliosidi come ST3GAL5-CDG e B4GALNT1-CDG sono estremamente rari e portano a gravi malattie neurodegenerative. I pazienti possono presentarsi con paraplegia spastica, grave ritardo intellettuale, epilessia e sintomi non neurologici tra cui displasia scheletrica, caratteristiche dismorfiche e pigmentazione anomala della pelle (59,60).
Mutazioni in molti geni all’interno della via biosintetica dell’ancora GPI causano una varietà di anomalie congenite multiple, disabilità intellettuale ed epilessia. Il difetto di biosintesi di GPI meglio caratterizzato, il deficit di PIGA legato all’X, si presenta con spasmi infantili con ipsaritmia, ipotonia, anomalie cerebrali multiple e dismorfismi facciali. I pazienti possono anche avere malattie cutanee, epatiche, cardiache e renali variabili (61-69). Alcune mutazioni all’interno di PIGA causano la malattia fenotipicamente distinta dell’emoglobinuria parossistica notturna (PNH), un disordine acquisito di insufficienza del midollo osseo (70,71).
Diagnosi
Quando si sospetta clinicamente una CDG, il primo passo è ordinare un test biochimico della CDG nel plasma o nel siero, compresi i test della CDT e degli N-glicani. L’analisi dei CDT e degli N-glicani nel siero può rilevare solo difetti di N-glicosilazione, quindi non sarebbe utile per differenziare difetti isolati di O-glicosilazione o di ancoraggio GPI. L’analisi delle isoforme della transferrina è stata originariamente ottenuta mediante focalizzazione isoelettrica della transferrina, poiché il fallimento della sintesi degli N-glicani causa una parziale carenza di acido sialico, che altera la carica sulla transferrina sierica e successivamente la sua migrazione catodica su un campo elettroforetico. Tuttavia, le analisi basate sulla spettrometria di massa della transferrina e degli N-glicani hanno ormai ampiamente sostituito la focalizzazione isoelettrica, identificando i cambiamenti specifici degli oligosaccaridi per massa e carica (72).
difetti di glicosilazione delle proteine N-collegate
I risultati CDT della transferrina nel siero sono riportati come rapporto tra transferrina mono-oligosaccaride/di-oligosaccaride, a-oligosaccaridi/di-oligosaccaridi transferrina, tri-sialo/di-oligosaccaridi transferrina, apolipoproteina CIII-1/apolipoproteina CIII-2, e rapporto apolipoproteina CIII-0/apolipoproteina CIII-2. Questi risultati quantitativi verranno anche con un’interpretazione del modello di risultati.
Un tipo I modello transferrina CDT è caratterizzato da bande di- e asialotransferrina aumentata, e indica difetti nella sintesi di N-glicano nel citosol o reticolo endoplasmatico. Un pattern di tipo II è caratterizzato da un aumento delle bande di- e asialotransferrina, e bande di tri- e/o monosialotransferrina, e indica difetti nell’elaborazione di N-glicani nell’apparato di Golgi (73).
Se viene rilevato un pattern di tipo I di transferrina CDT nel siero, il deficit di PMM2 o il deficit di MPI dovrebbe essere in prima linea nelle differenziazioni, poiché la PMM2-CDG è la CDG più comune e la MPI-CDG è curabile e potenzialmente mortale se non trattata. Per differenziare le diagnosi, dovrebbero essere intrapresi il profiling di N-glicani, il sequenziamento molecolare o i test enzimatici. La diagnosi di PMM2-CDG o MPI-CDG è confermata attraverso test molecolari che mostrano varianti patogene bialleliche in PMM2 o MPI, seguite dall’attività enzimatica PMM o MPI nei leucociti o nei fibroblasti se la patogenicità delle varianti genetiche è incerta. L’analisi degli N-glicani o l’analisi molecolare differenzierebbe la maggior parte delle ALG-CDG dal PMM2 o MPI-CDG (15).
Un pattern CDT della transferrina sierica di tipo II indica difetti del Golgi come il deficit di N-acetilglucosaminiltransferasi (GnT) II (CDG tipo IIA, MGAT2-CDG). L’analisi dell’isoforma dell’apolipoproteina CIII (Apo-CIII) è un test complementare per un profilo CDT di tipo II, poiché misura i difetti di glicosilazione O della mucina nell’apparato di Golgi. C’è una sensibilità limitata per CDT o Apo-CIII nel rilevare la CDG di tipo II. Così il profilo di N-glicano e O-glicano e il pannello molecolare o il sequenziamento dell’esoma dovrebbero essere intrapresi quando questi test clinici sono disponibili. I modelli di glicosilazione della transferrina possono normalizzarsi sporadicamente; pertanto, test ripetuti possono essere indicati in pazienti con alto indice di sospetto. Falsi positivi possono essere ottenuti in pazienti con crisi acuta di intolleranza ereditaria al fruttosio, galattosemia, malattia epatica acuta e alcune infezioni batteriche. Nessuno dei test biochimici di CDG può schermare tutti i CDG, quindi anche in presenza di risultati di screening normali, il test del pannello genico molecolare o il sequenziamento dell’esoma possono essere eseguiti per un forte sospetto clinico. Al contrario, la conferma biochimica e funzionale dei risultati genetici molecolari sono essenziali, poiché la maggior parte dei pazienti con CDG è portatrice di almeno una mutazione missense lieve e spesso nuova.
Difetti di glicosilazione O-linked e difetti combinati di glicosilazione N- e O-linked
La diagnosi si basa sul sequenziamento molecolare, poiché l’analisi delle isoforme della transferrina non rileverebbe difetti isolati di glicosilazione O. Difetti combinati di glicosilazione N- e O-linked possono essere rilevati tramite CDT, analisi ApoCIII e analisi di N-glicani e O-glicani nel plasma.
Difetti di glicosilazione dei lipidi e biosintesi di ancore GPI
La citometria a flusso dei granulociti del sangue misura l’espressione della superficie cellulare delle proteine ancorate a GPI come CD16 e CD24. L’analisi della citometria a flusso dei globuli bianchi o dei globuli rossi per alcune proteine della superficie cellulare ancorate a GPI è disponibile clinicamente come test per la PNH dovuta a mutazioni acquisite nel gene PIGA. Il test PNH può rivelare anomalie in altre carenze di ancoraggio GPI, ma la diagnosi si basa principalmente sull’analisi molecolare.
Analisi molecolare
Il più alto rendimento diagnostico per la CDG è il pannello di sequenziamento genico basato sulla prossima generazione o il sequenziamento dell’esoma clinico (CES). Il sequenziamento genetico controlla la sequenza nucleotidica, o l’ortografia “lettera”, dei geni per determinare se c’è un cambiamento che influenza la funzione del gene. Il genoma umano consiste di 3 milioni di nucleotidi, ma solo l’1-2% di questi, chiamati esoni, sono tradotti in un prodotto proteico funzionale. Il restante DNA non codificante intercalato tra gli esoni che non viene tradotto è chiamato introni (74). Il CES esamina quasi tutti gli esoni noti dei circa 20.000 geni del genoma umano, che rappresentano una minoranza del materiale genetico nei cromosomi, ma che molto probabilmente contengono varianti che causano malattie (patogene). CES può anche includere il sequenziamento del DNA mitocondriale (mtDNA), che interroga il piccolo DNA circolare extranucleare situato nei mitocondri che è esclusivamente ereditato dalla madre.
I possibili risultati per CES includono varianti positive, negative e di significato sconosciuto. Un risultato positivo significa che le varianti note che causano la malattia (cioè, patogene) sono identificate, dopo di che la diagnosi, la storia naturale, la prognosi, il rischio di recidiva e le opzioni di trattamento possono essere discusse. Un risultato negativo significa che non sono state identificate varianti patogene rilevabili. Varianti di significato sconosciuto (VUS) significa che anche se sono stati identificati cambiamenti genetici, non ci sono abbastanza informazioni sul cambiamento genetico specifico per sapere definitivamente se è causa di malattia. Ci si aspettano variazioni nel DNA di ogni individuo, quindi avere campioni parentali testati contemporaneamente per il confronto può aiutare il laboratorio e l’interpretazione clinica dei risultati. La resa diagnostica della CES è stimata al 30-35% e sta aumentando nel tempo con la scoperta dei geni e la conoscenza del genoma umano che continuano a progredire (75-77). Il CES è sempre più ordinato come test genetico di prima linea di scelta, dato il suo rapido tempo di risposta e il basso costo relativo per la quantità di informazioni genetiche analizzate. I limiti del CES includono la mancanza di una sensibilità del 100%, l’incapacità di rilevare alcuni tipi di cambiamenti genetici (ad esempio, delezioni, duplicazioni, ripetizioni trinucleotidiche, mutazioni introniche profonde o difetti di metilazione), e il fatto che una diagnosi può non fornire informazioni aggiuntive sulla malattia o cambiare la gestione.
Nella segnalazione del CES, varianti patogene rilevate incidentalmente in geni associati a condizioni genetiche ben note possono essere riportate come risultati secondari (78). Questo elenco di malattie raccomandate è curato dall’American College of Medical Genetics (ACMG). Il Genetic Information Nondiscrimination Act (GINA) è una considerazione importante quando si decide se optare o meno per l’apprendimento dei risultati accidentali (79). La GINA protegge gli individui dall’uso improprio delle informazioni genetiche nell’assicurazione sanitaria e nel lavoro, ma non nell’assicurazione sulla vita. La GINA protegge le seguenti informazioni genetiche: anamnesi familiare, test del portatore, test genetici prenatali, test di suscettibilità e predittivi, e analisi di tumori o altre valutazioni di geni, mutazioni o cambiamenti cromosomici.
Gestione
La gestione della CDG dipende in gran parte dai sintomi specifici dell’individuo. I sintomi ricorrenti nei pazienti con CDG includono mancata crescita, ritardo globale dello sviluppo, vomito, episodi simili all’ictus e anomalie scheletriche. Coagulopatia clinica o sub-clinica, endocrinopatia, epatopatia e difetti cardiaci sono anche comunemente visti. I test di laboratorio di base per stabilire l’estensione della malattia e il monitoraggio di routine sono raccomandati, specialmente per la PMM2-CDG. Questi includono test di funzionalità epatica, albumina del siero, test di funzionalità tiroidea incluso T4 libero, proteina C, proteina S, antitrombina III, fattore IX, analisi delle urine e gonadotropine del siero e ormone della crescita.
La diagnostica per immagini raccomandata include ecocardiogramma, ecografia renale, età ossea, esame oftalmologico per la valutazione di lente, retina, mobilità oculare e pressione intraoculare. Se non diversamente indicato, le vaccinazioni di routine sono raccomandate per adulti e bambini affetti da CDG. I titoli anticorpali devono essere ottenuti dopo la vaccinazione poiché i pazienti possono avere una risposta immunogenica subottimale. La reintegrazione profilattica dei fattori di coagulazione prima di qualsiasi procedura chirurgica può essere necessaria se esistono carenze al basale.
La valutazione genetica clinica dovrebbe essere intrapresa per discutere gli aspetti ereditari della CDG, così come stabilire una casa medica per questi pazienti complessi. La casa medica è tipicamente il servizio di genetica biochimica, anche se i dipartimenti di genetica, neurosviluppo o neurologia hanno anche servito in questa capacità se un servizio di genetica biochimica dedicato non è disponibile. Spesso è necessario l’invio di specialisti in gastroenterologia, ematologia, endocrinologia, supporto nutrizionale, logopedia, terapia occupazionale, fisica e alimentare, ortopedia e medicina riabilitativa.
Terapie mirate e prognosi
Il trattamento della maggior parte dei tipi di CDG è in gran parte di supporto, con poche eccezioni. La MPI-CDG è la più efficacemente trattabile di tutte le CDG. Il mannosio orale viene convertito in mannosio-6-fosfato dall’esochinasi intracellulare, aggirando così il blocco enzimatico e producendo il substrato carente. L’integrazione di mannosio inizia tipicamente a 1 g/kg di peso corporeo al giorno, diviso in 4-6 dosi al giorno. Mentre l’enteropatia proteica potenzialmente pericolosa per la vita è particolarmente sensibile al trattamento con mannosio, la malattia epatica nella MPI-CDG può continuare a progredire. I sintomi clinici migliorano rapidamente e la transferrina CDT si normalizza nel corso dei mesi, anche se la malattia epatica può continuare a progredire con il trattamento (45,80,81).
Si deve prestare attenzione nell’integrare il mannosio durante la gravidanza, poiché la somministrazione di mannosio in modelli di topi ipomorfi fosfomannosici isomerasi incinta ha portato a letalità embrionale e cecità nei loro cuccioli (82). Inoltre, il mannosio per via endovenosa è stato associato a una diminuzione della coscienza e a convulsioni, che si sono risolte con la somministrazione di glucosio (83).
Il trattamento della PMM2-CDG è in gran parte di supporto e basato sulla sintomatologia. Tuttavia, i prossimi studi clinici sulla terapia sostitutiva del substrato mannosio-1-fosfato sono attualmente in fase di sviluppo.
Per le altre CDG, sono stati studiati vari zuccheri semplici per via orale con lo scopo di migliorare teoricamente l’ipoglicosilazione. Il fucosio è stato provato per SLC35C1-CDG e il galattosio per PGM1-CDG e SLC35A2-CDG con risultati misti (84). Il D-galattosio a 1,0-2,5 g/kg/giorno (max 50 grammi) ha dimostrato di migliorare l’ipoglicemia, la coagulopatia e l’endocrinopatia in PGM1-CDG (85,86). Il galattosio ha anche dimostrato di migliorare l’endocrinopatia e la coagulopatia in TMEM165-CDG (87) e SLC39A8-CDG. Un notevole miglioramento clinico è stato riportato anche in pazienti SLC39A8-CDG con 15-20 mg/kg/giorno di MnSO4 (88). Sono in corso studi clinici per indagare l’utilità della N-acetilmannosamina (ManNAc) nella GNE-CDG (89), e diversi studi pre-clinici sono in corso per altre CDG (90).
Nonostante i progressi medici, esiste una significativa mortalità per i bambini con CDG entro il primo anno di vita per insufficienza multiorgano o infezioni gravi (91). I neonati con CDG possono presentare una malattia multiorgano fulminante, convulsioni intrattabili o grave ipoalbuminemia che progredisce fino all’anasarca. Alcuni pazienti rispondono alla diuresi aggressiva e alla sostituzione dell’albumina, mentre altri sono refrattari al trattamento. È stato dimostrato che il butirrato di sodio migliora il controllo delle crisi in CAD-CDG e PIGM-CDG (92). La dieta chetogenica ha anche dimostrato di diminuire la frequenza delle crisi in alcuni casi di PIGA-CDG (93). Durante gli episodi simili all’ictus, l’idratazione endovenosa e il mantenimento della glicemia normale possono essere utili mentre l’eziologia vascolare trombotica o emorragica sottostante viene esclusa.
Con l’avvento delle tecniche di genome-editing e una migliore comprensione del meccanismo delle malattie incluse nell’ombrello diagnostico della CDG, il futuro dello sviluppo terapeutico mirato rimane promettente.
Riconoscimenti
Vorremmo ringraziare Lynne Wolfe, ARNP e Donna Krasnewich, MD, PhD per averci fornito le foto cliniche ottenute come parte del Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). Vorremmo anche ringraziare Jenny Thies, MS, LGC per la sua esperienza nella consulenza genetica.
Finanziamento: IJ Chang è sostenuto dal National Institutes of Health T32GM007454.
Footnote
Conflitti di interesse: Gli autori non hanno conflitti di interesse da dichiarare.
Consenso informato: Il consenso informato scritto è stato ottenuto dai pazienti per la pubblicazione di questo manoscritto e qualsiasi immagine di accompagnamento.
- Jaeken J, Matthijs G. Disordini congeniti della glicosilazione. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Disturbi congeniti della glicosilazione: una famiglia di malattie in rapida espansione. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutazioni in PMM2 che causano disturbi congeniti di glicosilazione, tipo Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Disordini congeniti di glicosilazione: un aggiornamento sui difetti che interessano la biosintesi di oligosaccaridi legati al dolicholo. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Mancanza di equilibrio Hardy-Weinberg per la mutazione PMM2 più prevalente in CDG-Ia (disturbi congeniti della glicosilazione di tipo Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. Nomenclatura CDG: tempo per un cambiamento! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Risolvere i disturbi della glicosilazione: approcci fondamentali rivelano percorsi complicati. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Cosa c’è di nuovo nella CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Disturbi congeniti della glicosilazione. Parte I. Difetti di proteina N-glicosilazione. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Funzioni intracellulari di N-linked glicani. Scienza 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Risultati clinici, di laboratorio e molecolari e dati di follow-up a lungo termine in 96 pazienti francesi con PMM2-CDG (fosfomannomutasi 2-disordine congenito di glicosilazione) e revisione della letteratura. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. Un’indagine nazionale sulla PMM2-CDG in Italia: alta frequenza di una variante neurologica lieve associata alla mutazione L32R. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG in Sud Africa: Descrizione genotipo-fenotipo di cinque nuovi pazienti. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Diverse glicosiltransferasi sono elaborate in modo diverso per la secrezione, dimerizzazione e autoglicosilazione. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In: Mora-Montes H. Editor. Glicani: Biochimica, caratterizzazione e applicazioni Disturbi congeniti di glicosilazione. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Disturbi congeniti di glicosilazione: altre cause di ittiosi. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Manifestazioni cutanee in CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. Sistema IGF nei bambini con disturbi congeniti di glicosilazione. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. Un ampio spettro di presentazioni cliniche nei disturbi congeniti di glicosilazione I: una serie di 26 casi. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Crescita prepuberale nel disturbo congenito di glicosilazione tipo Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Caratteristiche cliniche e biochimiche del disturbo congenito di glicosilazione tipo Ic, il primo difetto riconosciuto reticolo endoplasmatico nella sintesi di N-glicano. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Disturbi congeniti della glicosilazione: revisione delle loro basi molecolari, presentazioni cliniche e terapie specifiche. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–una malattia metabolica ereditaria recentemente scoperta. Manifestazioni d’organo multiple, incidenza 1/80.000, difficile da trattare. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Grave ischemia miocardica transitoria causata da cardiomiopatia ipertrofica in un paziente con disordine congenito di glicosilazione tipo Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Difetti cardiaci conotrinali in tre pazienti con disordine congenito di glicosilazione tipo Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Accumulo di liquido pericardico e addominale nel disordine congenito di glicosilazione tipo Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Caratteristiche cliniche negli adulti con disturbi congeniti di glicosilazione di tipo Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Sindrome da glicoproteina carente di carboidrati. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Caratteristiche cliniche e molecolari del disordine congenito di glicosilazione in pazienti con pattern di sialotransferrina tipo 1 e origini etniche diverse. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modifica della proteina del mantello del virus Plum pox potyvirus migliora l’infezione virale. Virologia 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. pazienti scandinavi CDG-Ia: correlazione genotipo/fenotipo e origine geografica delle mutazioni fondatore. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 pazienti adulti francesi con PMM2-disturbo congenito della glicosilazione: risultato del fenotipo pediatrico classico e rappresentazione di un fenotipo tardivo. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Caratteristiche retiniche del disturbo congenito di glicosilazione PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Disturbo congenito di glicosilazione tipo Ia (CDG-Ia): spettro fenotipico del genotipo R141H/F119L. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Funzione tiroidea in PMM2-CDG: approccio diagnostico e gestione proposta. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Disturbo congenito di glicosilazione tipo Ia presentando come early-onset atassia cerebellare in un adulto. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Sviluppo mentale borderline in un disordine congenito di glicosilazione (CDG) tipo Ia paziente con coinvolgimento multisistemico (fenotipo intermedio). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. disordine congenito di glicosilazione tipo 1a: tre fratelli con un fenotipo neurologico lieve. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Nuovi disturbi nel metabolismo dei carboidrati: disturbi congeniti di glicosilazione e il loro impatto sul sistema endocrino. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Disturbo congenito di glicosilazione tipo Ia: eterogeneità nella presentazione clinica da insufficienza multiviscerale a ipoglicemia iperinsulinemica come sintomi principali in tre bambini con deficit di fosfomannomutasi. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinologia della sindrome da glicoproteina con deficit di carboidrati di tipo 1 dalla nascita all’adolescenza. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Funzione gonadica e ormoni glicoproteici nella sindrome da glicoproteina con deficit di carboidrati (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Lo spettro clinico del deficit di fosfomannosio isomerasi, con una valutazione del trattamento del mannosio per CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Complicazioni cardiache dei disturbi congeniti di glicosilazione (CDG): una revisione sistematica della letteratura. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Idrope fetalis ricorrente non immunitario associato alla sindrome della glicoproteina carente di carboidrati. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. deficit di fosfomannosio isomerasi: una sindrome da glicoproteina con presentazione epatico-intestinale. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Sindrome della glicoproteina carboidrato-deficiente tipo Ib. Carenza di fosfomannosio isomerasi e terapia con mannosio. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Ipoglicemia grave come sintomo di presentazione della sindrome della glicoproteina con deficit di carboidrati. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Ipoglicemia iperinsulinemica come segno di presentazione nel deficit di fosfomannosio isomerasi: Una nuova manifestazione della sindrome da glicoproteina carente di carboidrati trattabile con mannosio. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Scheda gene utilità clinica per: ALG6 difettoso disordine congenito di glicosilazione. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Muco delle vie aeree: Dalla produzione alla secrezione. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Esito clinico a lungo termine e variabilità fenotipica in calcinosi tumorale familiare iperfosfatemica e sindrome iperostosi iperfosfatemica causata da una nuova mutazione GALNT3; case report e revisione della letteratura. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Solfato di eparano nello sviluppo scheletrico, crescita e patologia: il caso delle esostosi multiple ereditarie. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Espandere lo spettro clinico del deficit di B4GALT7: mutazione omozigote p.R270C con effetto fondatore causa Larsen di Reunion Island sindrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Varie condizioni muscolo-scheletriche non infiammatorie. Calcolosi tumorale familiare iperfosfatemica (FGF23, GALNT3 e αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Ablazione del gene Galnt3 porta a basso circolante intatto fattore di crescita dei fibroblasti 23 (Fgf23) concentrazioni e iperfosfatemia nonostante una maggiore espressione Fgf23. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Una mutazione in un enzima ganglioside biosintetico, ST3GAL5, risultati in sale & sindrome di pepe, un disturbo neurocutaneo con alterata glicolipide e glicoproteina glicosilazione. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutazioni in B4GALNT1 (GM2 sintasi) alla base di un nuovo disturbo della biosintesi dei gangliosidi. Cervello 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. mutazioni PIGA causare encefalopatie epilettiche a esordio precoce e caratteristiche distintive. Neurologia. Neurologia 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalie-ipotonia-seizures sindrome è causato da una mutazione in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Mutazione del promotore ipomorfo in PIGM causa deficit ereditato di glicosilfosfatidilinositolo. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. mutazioni ipomorfe in PGAP2, codificando una proteina GPI-anchor-remodeling, causa autosomica-recessiva disabilità intellettuale. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. Il fenotipo di una mutazione germinale in PIGA: il gene mutato somaticamente in emoglobinuria parossistica notturna. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutazioni in PIGO, un membro della via di sintesi GPI-anchor, causa iperfosfatasia con ritardo mentale. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. Una nuova sindrome di disabilità intellettuale causata da deficit di ancoraggio GPI a causa di mutazioni omozigoti in PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutazioni nel gene glicosilfosfatidilinositolo PIGL causare la sindrome CHIME. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. Lo spettro genotipico e fenotipico del deficit di PIGA. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) è causato da mutazioni somatiche nel gene PIG-A. EMBO J 1994;13:110-7.
- Brodsky RA. Emoglobinuria parossistica notturna. Sangue 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. L’impatto della spettrometria di massa nella diagnosi dei disturbi congeniti di glicosilazione. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessiva cardiomiopatia dilatativa a causa di DOLK mutazioni risultati da anormale distroglicano O-mannosilazione. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distribuzioni di esoni e introni nel genoma umano. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing per la diagnosi di disturbi mendeliani. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Una migliore resa diagnostica rispetto ai pannelli di sequenziamento genico mirato suggerisce un ruolo per il sequenziamento dell’intero genoma come test genetico di primo livello. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. Il costo e la resa diagnostica del sequenziamento dell’esoma per i bambini con sospetti disturbi genetici: uno studio di benchmarking. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Raccomandazioni per la segnalazione di risultati secondari nel sequenziamento dell’esoma e del genoma clinico, aggiornamento 2016 (ACMG SF v2.0): una dichiarazione politica dell’American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Correnti nell’etica contemporanea. GINA, l’ADA e la discriminazione genetica nel lavoro. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Trombo-embolia ricorrente in un bambino con un disturbo congenito di glicosilazione (CDG) tipo Ib e trattamento con mannosio. Pediatr Hematol Oncol 2008;25:762-8.
- Menzione K, Lacaille F, Valayannopoulos V, et al. Sviluppo della malattia epatica nonostante il trattamento con mannosio in due pazienti con CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplementi inducono letalità embrionale e cecità in topi ipomorfi fosfomannoso isomerasi. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Convulsioni e stupor durante la terapia endovenosa mannose in un paziente con sindrome CDG tipo 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. La supplementazione di glucosio nel deficit di adesione dei leucociti di tipo II. Sangue 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Carenza ereditaria di glicosilfosfatidil inositolo: una CDG curabile. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Supplemento di D-galattosio orale in PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. La supplementazione di galattosio in pazienti con TMEM165-CDG salva i difetti di glicosilazione. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. Deficit SLC39A8: correzione biochimica e importante miglioramento clinico dalla terapia con manganese. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Terapie orali di monosaccaridi per invertire l’iposialilazione renale e muscolare in un modello murino di miopatia GNE. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: Dal banco al letto. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Disturbi della glicosilazione umana. Marino PA, editore. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Dieta chetogenica – un nuovo trattamento per l’encefalopatia epilettica precoce dovuta al deficit di PIGA. Brain Dev 2016;38:848-51.
.