- Översikt
- Epidemiologi
- Biokemisk klassificering och nomenklatur
- Genetik
- Patofysiologi
- N-länkade proteinglykosyleringsdefekter
- O-länkade glykosyleringsdefekter och kombinerade N- och O-länkade glykosyleringsdefekter
- Lipidglykosylering och defekter i biosyntesen av GPI-ankrar
- Kliniska manifestationer
- N-kopplade proteinglykosyleringsdefekter
- PMM2-CDG (CDG-Ia, PMM2-brist)
- MPI-CDG (CDG-Ib, mannosefosfatisomerasbrist)
- ALG6-CDG (glukosyltransferas 1-brist)
- O-länkade glykosyleringsdefekter och kombinerade N- och O-länkade glykosyleringsdefekter
- Defekter i lipidglykosylering och GPI-ankersbiosyntes
- Diagnos
- N-länkade proteinglykosyleringsdefekter
- O-kopplade glykosyleringsdefekter och kombinerade N- och O-kopplade glykosyleringsdefekter
- Defekter i lipidglykosylering och GPI-ankringsbiosyntes
- Molekylär analys
- Hantering
- Målinriktade terapier och prognos
- Acknowledgements
- Fotnot
Översikt
Glykosylering är en process där sockerrester läggs till proteiner och lipider i olika cellulära vägar. Medfödda glykosyleringsstörningar (Congenital disorders of glycosylation, CDG) är en genetiskt och kliniskt heterogen grupp av över hundra sjukdomar som orsakas av defekter i olika steg längs glykansyntesen eller modifieringsvägarna. De flesta av dessa monogena sjukdomar är autosomalt recessiva i arv, men autosomalt dominanta och X-bundna former har också beskrivits.
CDG uppvisar vanligen multisystemiska manifestationer, oftast utvecklingsförsening, bristande tillväxt, hypotoni, neurologiska avvikelser, hepatopati och koagulopati. Drabbade individer kan också uppvisa ögon-, hud- och hjärtsjukdomar samt dysmorfism i ansiktet. Även om neurologiska förändringar och kognitiva förseningar ses hos majoriteten av de drabbade individerna finns det vissa fall och även typer som inte har neurologiska manifestationer. Med tanke på den breda kliniska och genetiska etiologin för CDG bygger den kliniska diagnosen på ett högt index av misstankar vid multisystemisk sjukdom.
Serumanalys av kolhydratbristande transferrin (CDT) är det första screeningstestet hos patienter med misstänkt CDG, men är begränsat i upptäckt till N-glykosyleringsdefekter med sialinsyrabrist. Nästa test omfattar analys av dolichol-länkade glykaner och genetisk testning. Tidig diagnos av denna grupp av exponentiellt växande sjukdomar är viktig, eftersom vissa CDG kan behandlas. Behandling av glykosyleringsdefekter är huvudsakligen stödjande, även om riktade terapier finns tillgängliga för MPI-CDG, SLC35C1-CDG, PIGM-CDG och PGM1-CDG. Detaljer om dessa behandlingar finns under avsnittet ”Riktade terapier och prognos” nedan. Fokus i denna genomgång kommer att ligga på de vanligaste typerna av CDG med igenkännbara fenotyper eller behandlingar, med målgruppen primärvårdspersonal.
Epidemiologi
Incidensen och prevalensen av alla typer av CDG sammantaget har inte fastställts på ett välgrundat sätt, även om patienter har rapporterats över hela världen från nästan alla etniska bakgrunder och båda könen drabbas lika mycket. Den uppskattade prevalensen i europeiska och afroamerikanska populationer är 1/10 000 baserat på bärarfrekvenser av kända patogena varianter i 53 gener (1-4). Prevalensen för den vanligast diagnostiserade CDG, PMM2-CDG, varierar mellan 1/20 000 i holländska befolkningar och 1/77 000 i Estland baserat på isolerade rapporter (5,6). Hittills har färre än 100 fall rapporterats för de flesta CDG-typer.
Biokemisk klassificering och nomenklatur
I stort sett klassificeras CDG för närvarande i fyra kategorier – (I) N-länkad glykosylering, (II) O-länkad glykosylering, (III) kombinerad N- och O-länkad/multiplikatorglykosylering och (IV) defekter i biosyntesen av lipid- och glykosylfosfospatidylinositol- (GPI-) ankare.
Den N-länkade proteinglykosyleringsdefekten PMM2-CDG, tidigare känd som CDG typ Ia, var den första CDG som rapporterades av Jaeken 1980 och är fortfarande den överlägset vanligaste CDG:n hittills (7). PMM2-CDG kallades ursprungligen för ”carbohydrate-deficient glycoprotein syndrome” på grund av flera serumglykoproteinavvikelser som sågs genom isoelektrisk fokusering av serumtransferrin hos drabbade individer. Historiskt sett klassificerades CDG efter mönster för analys av transferrinisoformer – typ I-mönster tillskrevs dolichol-länkade glykansamlings- och överföringsfel som lokaliserades till cytoplasman eller ER, och typ II-mönster tillskrevs bearbetningsfel i Golgiapparaten. Från denna förgreningspunkt namngavs CDG:erna sedan alfabetiskt i den ordning de upptäcktes.
Med tillkomsten av utbredd molekylär diagnostik uppdaterades CDG-nomenklaturen 2008 för att specificera den molekylära etiologin för sjukdomen, vilket avspeglar den exponentiella tillväxten av banor och störningar som inte passar in i de tidigare etablerade dikotomiska kategorierna. För närvarande betecknas CDG-nomenklaturen av det drabbade gennamnet (icke-vitaliserat, gennamn på www.genenames.org), följt av -CDG (t.ex. PMM2-CDG) (8).
Genetik
Den stora majoriteten av de medfödda glykosyleringsstörningarna nedärvs autosomalt recessivt, med en mutation som nedärvs från var och en av de asymtomatiska föräldrarna (bärare). Molekylär testning, vanligtvis med nästa generations sekvenseringsmetoder, är nödvändig för att fastställa en genetisk diagnos. Föräldratest för den kända varianten kan bekräfta arv kontra de novo-förekomst. Vid autosomal recessiv nedärvning är återfallsrisken för syskon och varje graviditet hos en drabbad individ 25 % för att vara drabbad, 50 % för att vara asymtomatisk bärare och 25 % för att vara opåverkad.
En handfull CDG har autosomal dominant nedärvning (N-länkad: GANAB-CDG, PRKCSH-CDG; O-länkad: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Färre är X-länkade (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). De flesta dominanta och vissa X-kopplade former av CDG beror på de novo-mutationer. Specifika sjukdomar och gener beskrivs nedan i avsnittet ”patofysiologi”.
Mutationsdata för alla publicerade gener för CDG finns tillgängliga på Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Information om specifika genvarianter finns på Leiden Open Variation Database med integrerade in silico patogenicitetsverktyg (http://www.lovd.nl/3.0/home). Kliniska sammanfattningar för specifika gener finns på Online Mendelian Inheritance in Man (http://www.omim.org/) eller i mer begränsad omfattning på GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Med tanke på det lilla antalet drabbade patienter för de flesta CDG-subtyper är genotyp-fenotypkorrelationen svår att fastställa.
Patofysiologi
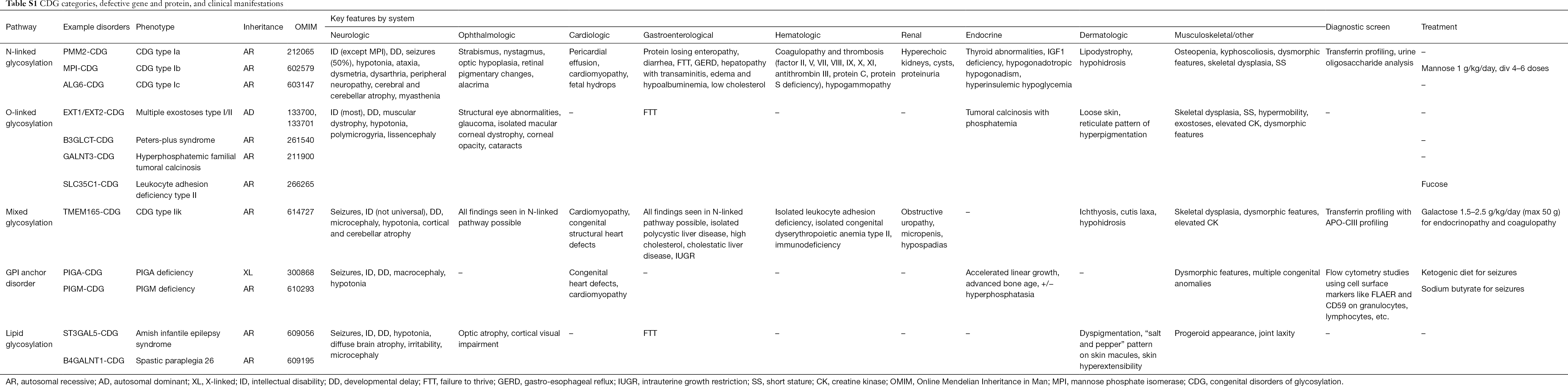
Över 130 typer av CDG har hittills rapporterats (9,10). Med tanke på den allestädes närvarande glykosyleringsvägarna är CDG extremt varierande i sin biokemiska patogenes. Många proteiner och lipider (dvs. sfingolipider och glykolipider) genomgår glykosylering med monosackarider och/eller oligosackarider, gemensamt kallade glykaner, i olika cellulära kompartment. Deras subcellulära placering är varierande, men de flesta defekter uppstår i ER- eller Golgi-apparaten. Kliniska särdrag och genetisk etiologi för vanligare CDG per väg sammanfattas i tabell S1.
Av proteiner beskrivs glykaner genom sin koppling till polypeptidkedjan – N-glykaner är knutna till amidgruppen i asparagin (Asn) medan O-glykaner är knutna till hydroxylgruppen i antingen serin eller threonin. N-glykansyntesen kräver en stegvis uppbyggnad av nukleotidlänkade sockerarter i cytosolen, sammansättning i det endoplasmatiska retikulumet och bearbetning i Golgiapparaten. O-glykansyntesen kräver däremot sammansättning men ingen bearbetning, varför defekter i O-glykosylering främst förekommer i Golgiapparaten.
N-länkade proteinglykosyleringsdefekter
N-glykosylering innebär kovalent bindning av kolhydratstrukturer till sidokedjans amidgrupp av Asn-rester inom en konsensus Asn-X-Ser/Thr-acceptorplats, translokation av substratpolypeptiden till det endoplasmatiska retikulumet för remodellering och ytterligare modifiering av N-glykan-kedjan inom Golgi (11,12). Defekter någonstans längs syntes-, sammansättnings- och bearbetningsvägen kan leda till klinisk sjukdom.
PMM2-CDG orsakas av patogena varianter i genen för fosfomannomutas 2 (PMM2), vilket leder till brist på PMM2-enzymet som katalyserar den cytosoliska omvandlingen av mannose-6-fosfat till mannose-1-fosfat i det andra steget av guanosindifosfat-(GDP)-mannossyntesen. De flesta patienter har sammansatta heterozygota patogena missense-mutationer (www.lovd.nl/PMM2). Den vanligaste återkommande patogena varianten p.Arg141His finns hos cirka 40 % av de drabbade individerna av europeisk härstamning, och p.Phe119Leu finns också ofta i norra Europa (1). Genotyp-fenotyp korrelationer har rapporterats för PMM2-CDG (3,13,14).
MPI-CDG är en autosomalt recessiv sjukdom som orsakas av patogena varianter i mannosofosfatisomeras (MPI) genen som leder till bristfälligt fosfomannosisomeras (MPI). MPI katalyserar normalt det första steget i GDP-mannos-syntesen (dvs. omvandlingen av fruktos-6-fosfat till mannos-6-fosfat), men fruktos-6-fosfat ackumuleras inte intracellulärt eftersom det också kan metaboliseras genom den glykolytiska vägen. Även om MPI-CDG biokemiskt liknar PMM2-CDG, orsakar MPI-CDG därför inte lika betydande neurologisk och multisystemisk påverkan. CDT är också det screeningprov som är det bästa valet för MPI-CDG, som visar ett typ 1-mönster. Diagnosen kan sedan bekräftas molekylärt eller genom fibroblast-/leukocyt-MPI-aktivitet.
ALG6-CDG är en recessiv sjukdom som orsakas av mutationer i ALG6, vilket leder till onormal bindning av tre glukosmolekyler till dolichol-länkade mannosemellanprodukter och nedströms hypoglykosylering av serumglykoproteiner (15).
O-länkade glykosyleringsdefekter och kombinerade N- och O-länkade glykosyleringsdefekter
O-glykosylering omfattar den stegvisa tillsättningen av kolhydratkedjor till serin-, threonin- och hydroxylysinrester i proteiner av glykosyltransferaser i Golgiapparaten (16). Flera typer av O-länkade glykaner har förknippats med sjukdomar hos människor, namngivna efter det första sockret som är knutet till aminosyraresten (17).
Lipidglykosylering och defekter i biosyntesen av GPI-ankrar
GPI-ankrar är glykolipider som genomgår sekventiell sammansättning i det endoplasmatiska retikulumet och modifieringar inom Golgi. GPI-ankersbiosyntesstörningar som beror på enzymbrister är namngivna i alfabetisk ordning efter upptäcktsordning och inte kronologiskt efter steg i sammansättningen. När de väl är syntetiserade finns GPI-ankrarna på plasmamembranen och binder hundratals proteiner på cellytan, vilket ger dem en mängd cellulära funktioner. De flesta av dessa sjukdomar är autosomalt recessiva med det anmärkningsvärda undantaget X-bunden PIGA-brist.
Kliniska manifestationer
Med tanke på den allestädes närvarande glykosyleringsvägarna kan i stort sett alla organsystem vara involverade i CDG, även om de flesta fall innefattar neurologiska avvikelser. En del CDG uppvisar ichtyos, däribland MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (figur 1) och PIGL-CDG (18,19). Nästan alla CDG uppvisar multisystemisk sjukdom inom de första levnadsåren, förutom att vissa påverkar endast ett enda organsystem (t.ex, näthinnan i DHDDS-CDG, den neuromuskulära korsningen i ALG2-CDG, ALG14-CDG, CFPT1-CDG, hjärnan i ST3GAL3-CDG, TUSC3-CDG, huden eller skelettmuskulaturen i POGLUT1-CDG, POFUT1-CDG, brosk i EXT1/EXT2-CDG, levern i TMEM199-CDG, de röda blodkropparna i SEC23B-CDG). Debutåldern och svårighetsgraden kan variera från neonatal dödlig till nästan asymtomatisk vuxen ålder, och alla permutationer däremellan. Den vanligaste rapporterade symtomkonstellationen omfattar utvecklingsförsening, bristande tillväxt, hypotoni, neurologiska avvikelser, hypoglykemi och varierande lever-, ögon-, hud-, gastrointestinala, immunologiska, skelett- och koagulationsavvikelser (19).
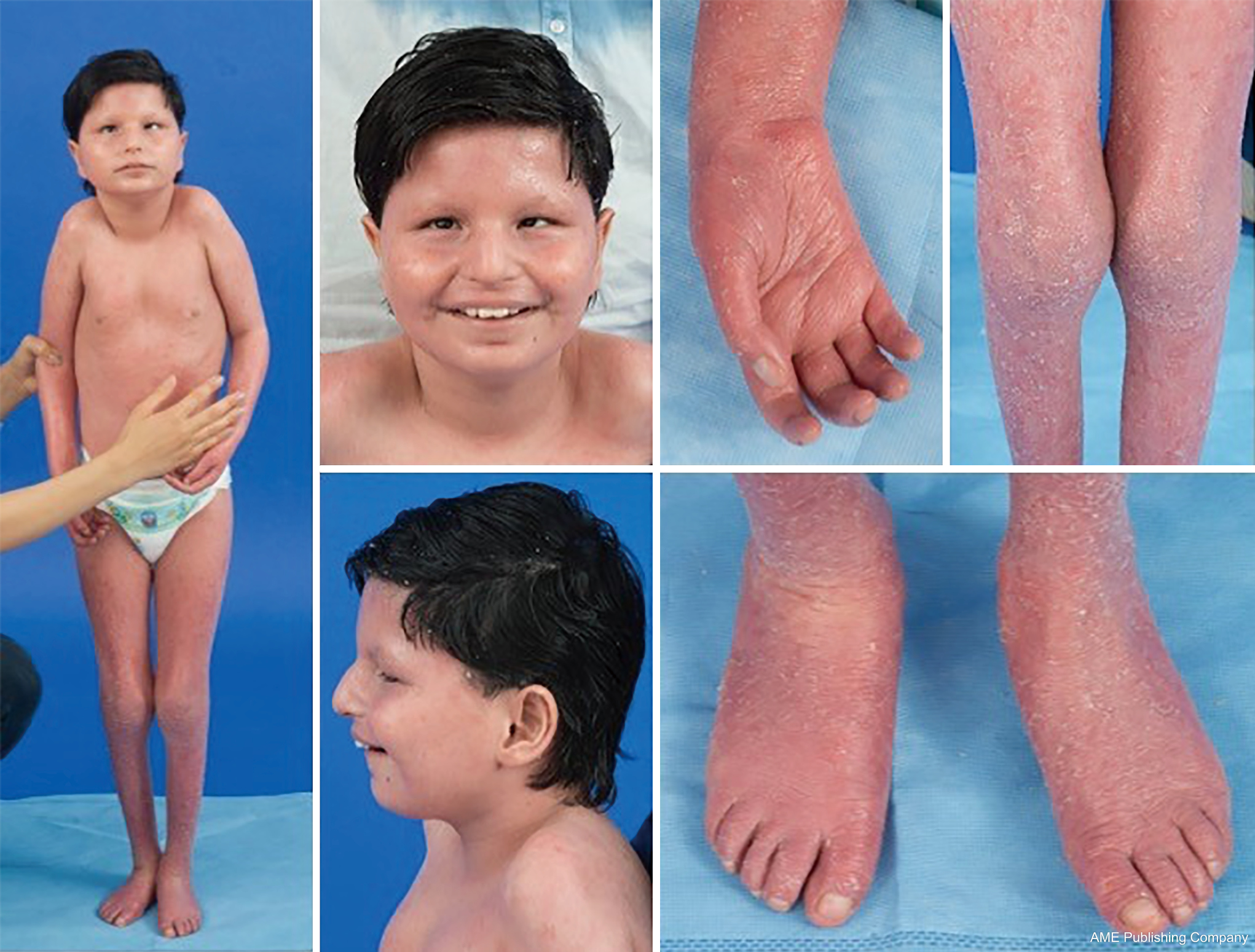
Den fullständiga fenotypen för många CDG-subtyper är fortfarande inte helt klarlagd på grund av de sällsynta rapporterade fallen. Därför bör CDG övervägas i alla sammanhang med multisystemisk sjukdom, särskilt i fall med en neurologisk komponent eller ospecifik utvecklingsfördröjning med oklar etiologi.
Och även om patofysiologin för multisymptom fortfarande inte är klarlagd har förhållandet mellan vissa glykosyleringsvägar och specifika kliniska symtom klargjorts. Till exempel beror den bristande tillväxt som ses i många typer av CDG på hypoglykosylering och försämrad bildning av flera glykoproteiner inom insulintillväxtvägen, inklusive IGF-1, ALS och IGFBP-3 (20). I takt med att vi bättre förstår denna grupp av komplexa sjukdomar, upptäcks CDG allt oftare hos individer med svårfångade diagnoser. De organsystem som är involverade i olika CDG sammanfattas i tabell S1. Vi kommer att diskutera de kliniska egenskaperna hos de vanligaste formerna och former med riktade behandlingar av CDG nedan.
Fullständig tabell
N-kopplade proteinglykosyleringsdefekter
Som den vanligaste diagnostiserade CDG:n, är fenotypen av N-kopplade glykosyleringsstörningar ofta utropad som den klassiska presentationen. Det fenotypiska spektrumet av CDG är dock mycket varierande, och många CDG kanske inte uppvisar stereotypa symtom som förknippas med PMM2-CDG.
PMM2-CDG (CDG-Ia, PMM2-brist)
PMM2-CDG är den vanligaste CDG:n, med över 700 rapporterade fall i världen. Den kännetecknas av multisystemisk allvarlig sjukdom i spädbarnsåldern, neurologisk sjukdom och utvecklingsförsening i barndomen och/eller stabil intellektuell funktionsnedsättning i vuxen ålder (21,22).
Under spädbarnsåldern uppvisar PMM2-CDG neurologiska avvikelser som typiskt sett uppträder strax efter födseln, nämligen strabism och onormala ögonrörelser, hypoplasi i lillhjärnan, hypotonin, psykomotorisk retardation, ataxi, hypotonin och hyporeflexi. Spädbarn kan också ha leversjukdom, nefrotiskt syndrom och njurcystor, perikardutgjutning och hypertrofisk kardiomyopati, sviktande tillväxt och multiorgansvikt som leder till döden under det första levnadsåret hos upp till 20 % av de drabbade individerna (21,23-28).
En konstellation av dysmorfiska drag har beskrivits hos patienter med PMM2-CDG (figurer 2,3). Dessa inkluderar ett hypoplastiskt cerebellum, dysmorfism i ansiktet (dvs. stora, dysplastiska öron), inverterade bröstvårtor och onormal fördelning av fettvävnad över skinkor eller suprapubisk region som kan försvinna med åldern (14,21,29-32). Patienterna har beskrivits ha en utåtriktad och glad framtoning. Presentationen är mycket varierande, även om strabism kan ses hos mer än 70 % av de drabbade patienterna (21,23,33-35). Inverterade bröstvårtor och onormala fettkuddar ses hos cirka 25-50 % av patienterna (36).

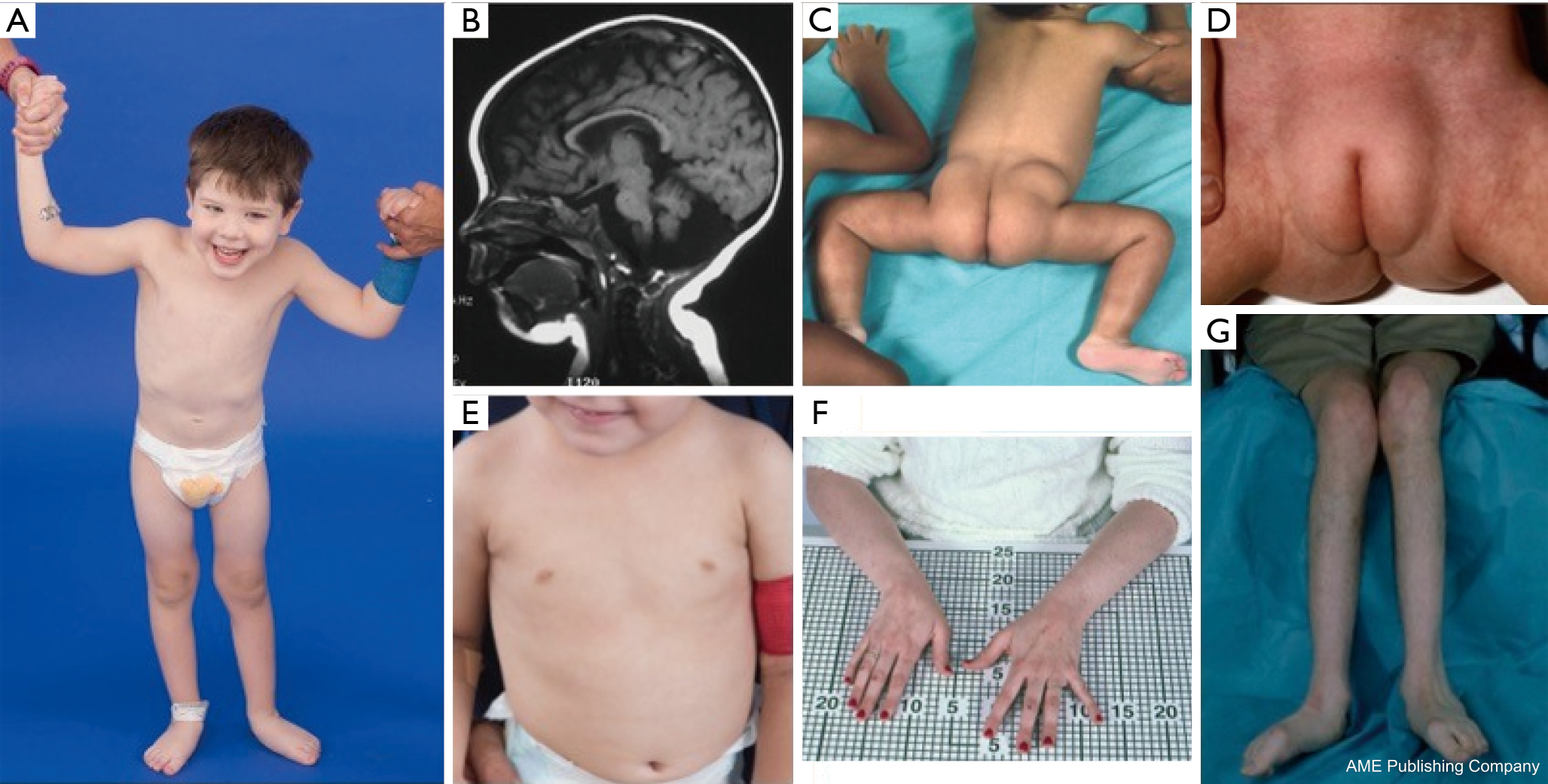
I barndomen kan drabbade individer utveckla retinitis pigmentosa, stroke-liknande episoder och kramper, förseningar i tal och motorik samt perifer neuropati. Konstitutionellt sett har patienterna vanligen misslyckad utveckling på grund av matnings- och GI-avvikelser och global utvecklingsfördröjning. Förhöjda levertransaminaser utan klinisk konsekvens kan observeras, vilka vanligtvis normaliseras vid 5 års ålder med tillfälliga fluktuationer vid sjukdom (21,24). Leverbiopsier är sällan indicerade vid CDG om man inte misstänker leverfibros (1). Klinisk hypotyreos är sällsynt, men patienter med CDG bör få sina sköldkörtelhormoner och fritt T4 mätt, vilket kan visa lågt sköldkörtelbindande globulin (TBG) och övergående förhöjningar av sköldkörtelstimulerande hormon (TSH) (37). Lever- och gallgångsmissbildningar har inte rapporterats hos PMM2-CDG-patienter.
Vuxna med PMM2-CDG kan leva fram till sjunde eller åttonde decenniet med stabil kognitiv fördröjning, perifer neuropati och progressiv thorakal och spinal kyfoskolios med osteopeni eller osteoporos (34). Cerebellär ataxi är ett alltmer erkänt symptom tillsammans med multisystemisk involvering (38-40). Endokrina avvikelser inklusive hyperprolaktinemi, frisättning av tillväxthormon med hyperglykemi, insulinresistens och hyperinsulinemisk hypoglykemi (41,42). Hos drabbade kvinnor kan hypogonadotropisk hypogonadism leda till utebliven sekundär sexuell utveckling eller avsaknad av äggstockar (41,43,44). Patienterna kan ha en ökad risk för trombos på grund av minskade koagulationsfaktorer i serum, inklusive faktorerna IV, IX och XI, antitrombin III, protein C och protein S (29).
MPI-CDG (CDG-Ib, mannosefosfatisomerasbrist)
MPI-CDG är unik eftersom drabbade patienter har liten eller ingen neurologisk involvering och vissa manifestationer av sjukdomen kan behandlas med oral mannos (2). Symtomen är huvudsakligen hepatisk-intestinala utan dysmorfiska drag eller kognitiva förseningar. Patienterna uppvisar vanligtvis återkommande kräkningar, betydande hypoglykemi, bristande tillväxt, potentiellt livshotande proteinförstörande enteropati, leverfibrotiska förändringar och dilatation av gallgångarna (45-51). Patienterna löper ökad risk för trombotiska händelser på grund av låga serumkoncentrationer av protein C och S och antitrombin III.
ALG6-CDG (glukosyltransferas 1-brist)
ALG6-CDG är den näst vanligaste N-glykosyleringsdefekten som kännetecknas av liknande men mildare fenotyp än PMM2-CDG. Patienter med ALG6-CDG har bristande tillväxt, utvecklingsförsening, hypotoni, kramper, strabism, ataxi, koagulopati och dysmorfism i ansiktet (t.ex. lågt sittande öron, hypertelorism och makroglossi). I likhet med MPI-CDG kan de också ha proteinförlorande enteropati. Dessutom kan drabbade patienter ha skelettmissbildningar, inklusive brakydaktyli och fingermissbildningar samt skolios. Affekterade patienter har vanligtvis inte typiskt sett retinitis pigmentosa eller cerebellär hypoplasi (52).
O-länkade glykosyleringsdefekter och kombinerade N- och O-länkade glykosyleringsdefekter
På grund av den betydande förekomsten av O-glykaner i mucininnehållande proteiner, inklusive glykosaminoglykaner (GAG) och epitelytor (53), leder störningar i GAG-syntesen typiskt sett till skelettdysplasier eller bindvävssjukdomar. Drabbade patienter kan uppvisa muskuloskeletala, hud- och ledavvikelser (t.ex. ledlaxitet, multipla exostoser, kondro/osteosarkom) utöver neurologiska symtom (54-56). N-acetylgalaktosaminyltransferas 3 (GALNT3) O-glykosylerar till exempel det fosfatursänkande hormonet FGF23, vilket förhindrar proteolytisk klyvning och möjliggör intakt sekretion. GALNT3-brist leder till familjär tumörkalcinos, som kännetecknas av hyperfosfatemi och ektopiska förkalkningar (57,58).
Defekter i lipidglykosylering och GPI-ankersbiosyntes
Glykosfosfingolipider och deras sialylerade derivat, gangliosider, uttrycks huvudsakligen av neuroner. Defekter i nedbrytningen av gangliosider leder till ackumulering och de välkaraktäriserade lysosomala lagringssjukdomarna. I motsatt riktning är defekter i gangliosidbiosyntesen, såsom ST3GAL5-CDG och B4GALNT1-CDG, ytterst sällsynta och leder till allvarliga neurodegenerativa sjukdomar. Patienterna kan uppvisa spastisk paraplegi, allvarlig intellektuell försening, epilepsi och icke-neurologiska symtom som skelettdysplasi, dysmorfiska drag och onormal hudpigmentering (59,60).
Mutationer i många gener inom GPI-ankersyntesvägen orsakar en mängd olika multipla medfödda anomalier, intellektuell funktionsnedsättning och epilepsi. Den bäst karakteriserade GPI-biosyntesdefekten, X-bunden PIGA-brist, visar sig genom spasmer hos barn med hypsarytmi, hypotoni, flera hjärnanormaliteter och ansiktsdysmorfism. Patienterna kan också ha varierande hud-, lever-, hjärt- och njursjukdomar (61-69). Vissa mutationer inom PIGA orsakar den fenotypiskt distinkta sjukdomen paroxysmal nattlig hemoglobinuri (PNH), en förvärvad sjukdom med benmärgssvikt (70,71).
Diagnos
När en CDG misstänks kliniskt är det första steget att beställa biokemiska CDG-tester i plasma eller serum, inklusive CDT- och N-glykanstester. CDT- och N-glykananalyser i serum kan endast upptäcka N-glykosyleringsdefekter, varför de inte skulle vara användbara för att differentiera isolerade O-glykosylerings- eller GPI-ankringsdefekter. Analys av transferrinisoformer gjordes ursprungligen genom isoelektrisk fokusering av transferrin, eftersom misslyckad N-glykansyntes orsakar partiell brist på sialinsyra, vilket ändrar serumtransferrins laddning och därefter dess katodala migration på ett elektroforetiskt fält. Masspektrometribaserad analys av transferrin och N-glykan har nu dock till stor del ersatt isoelektrisk fokusering genom att identifiera specifika förändringar i oligosackarider genom massa och laddning (72).
N-länkade proteinglykosyleringsdefekter
Serumtransferrin CDT-resultat rapporteras som förhållandet mellan mono-oligosackarid/di-oligosackaridtransferrin, a-oligosackarid/di-oligosackaridtransferrin, tri-sialo/di-oligosackaridtransferrin, apolipoprotein CIII-1/apolipoprotein CIII-2 och apolipoprotein CIII-0/apolipoprotein CIII-2. Dessa kvantitativa resultat kommer också med en tolkning av fyndmönstret.
En transferrin-CDT med typ I-mönster kännetecknas av ökade di- och asialotransferrinband och indikerar defekter i N-glykansyntesen i cytosolen eller det endoplasmatiska retikulumet. Ett typ II-mönster kännetecknas av ökade di- och asialotransferrinband och tri- och/eller monosialotransferrinband och indikerar defekter i N-glykanbearbetningen i Golgi-apparaten (73).
Om ett serumtransferrin-CDT-mönster av typ I påvisas bör PMM2-brist eller MPI-brist stå i förgrunden av differentieringarna, eftersom PMM2-CDG är den vanligaste CDG:n och MPI-CDG:n är behandlingsbar och potentiellt dödlig om den lämnas obehandlad. För att skilja mellan diagnoserna bör N-glykanprofilering, molekylär sekvensering eller enzymatisk testning genomföras. Diagnosen PMM2-CDG eller MPI-CDG bekräftas genom molekylär testning som visar bialleliska patogena varianter i PMM2 eller MPI, följt av PMM- eller MPI-enzymaktivitet i leukocyter eller fibroblaster om de genetiska varianternas patogenicitet är osäker. N-glykananalys eller molekylär analys skulle differentiera majoriteten av ALG-CDG från PMM2 eller MPI-CDG (15).
Ett typ II serumtransferrin CDT-mönster indikerar Golgi-defekter såsom N-acetylglukosaminyltransferas (GnT) II-brist (CDG typ IIA, MGAT2-CDG). Analys av apolipoprotein CIII (Apo-CIII) isoform är ett kompletterande test för en CDT-profil av typ II, eftersom den mäter mucintyp O-glykosyleringsdefekter i Golgiapparaten. Det finns en begränsad känslighet för CDT eller Apo-CIII när det gäller att upptäcka CDG av typ II. Därför bör N-glykan- och O-glykanprofilering och molekylär panel- eller exomsekvensering genomföras när dessa kliniska tester är tillgängliga. Transferrin-glykosyleringsmönster kan normaliseras sporadiskt; därför kan upprepade tester vara indicerade hos patienter med högt index av misstanke. Falskt positiva resultat kan erhållas hos patienter med akut kris av ärftlig fruktosintolerans, galaktosemi, akut leversjukdom och vissa bakterieinfektioner. Inget av de biokemiska CDG-testerna kan screena för alla CDG:er, så även i närvaro av normala screeningresultat kan molekylär genpanelstestning eller exomsekvensering utföras vid stark klinisk misstanke. Omvänt är biokemisk och funktionell bekräftelse av molekylärgenetiska fynd också väsentlig eftersom majoriteten av patienterna med CDG bär på minst en mild och ofta ny missense-mutation.
O-kopplade glykosyleringsdefekter och kombinerade N- och O-kopplade glykosyleringsdefekter
Diagnostiken är beroende av molekylär sekvensering, eftersom transferrin-isoformanalyser inte skulle kunna upptäcka isolerade O-glykosyleringsdefekter. Kombinerade N- och O-länkade glykosyleringsdefekter kan påvisas genom CDT, ApoCIII-analys och plasmaanalys av N-glykaner och O-glykaner.
Defekter i lipidglykosylering och GPI-ankringsbiosyntes
Flödescytometri av blodgranulocyter mäter cellytans uttryck av GPI-förankrade proteiner, såsom CD16 och CD24. Flödescytometrianalys av vita blodkroppar eller röda blodkroppar för vissa GPI-förankrade cellytproteiner finns tillgängliga kliniskt som ett test för PNH på grund av förvärvade mutationer i PIGA-genen. PNH-testet kan avslöja avvikelser vid andra GPI-ankringsbrister, men diagnosen bygger oftast på molekylär analys.
Molekylär analys
Den högsta diagnostiska avkastningen för CDG är nästa generations baserad gensekvenseringspanel eller klinisk exomsekvensering (CES). Genetisk sekvensering korrekturläser nukleotidsekvensen, eller bokstavsskrivningen, av gener för att avgöra om det finns en förändring som påverkar genens funktion. Det mänskliga genomet består av 3 miljoner nukleotider men endast 1-2 % av dessa, så kallade exoner, översätts till en funktionell proteinprodukt. Det återstående icke-kodande DNA som ligger mellan exoner och som inte översätts kallas introner (74). CES undersöker nästan alla kända exoner av de cirka 20 000 generna i det mänskliga genomet, som utgör en minoritet av det genetiska materialet i kromosomerna men som med största sannolikhet innehåller sjukdomsframkallande (patogena) varianter. CES kan också omfatta sekvensering av mitokondrie-DNA (mtDNA), som förhör det lilla, extranukleära, cirkulära DNA som finns i mitokondrierna och som uteslutande är nedärvt från modern.
De möjliga resultaten för CES omfattar positiva, negativa och varianter av okänd betydelse. Ett positivt resultat innebär att kända sjukdomsorsakande (dvs. patogena) varianter identifieras, varefter diagnos, naturalhistoria, prognos, återfallsrisk och behandlingsalternativ kan diskuteras. Ett negativt resultat innebär att inga påvisbara patogena varianter har identifierats. Varianter av okänd betydelse (även kallade VUS) innebär att även om genetiska förändringar identifierades finns det inte tillräckligt med information om den specifika genetiska förändringen för att definitivt veta om den är sjukdomsorsakande. Variationer i varje individs DNA är förväntade, varför det kan underlätta laboratoriets och den kliniska tolkningen av resultaten att låta föräldraproverna testas samtidigt för jämförelse. Det diagnostiska utbytet av CES uppskattas till 30-35 % och ökar med tiden i takt med att genupptäckten och kunskapen om det mänskliga genomet fortsätter att utvecklas (75-77). CES beställs allt oftare som förstahandsval för bred genetisk testning med tanke på dess snabba handläggningstid och låga relativa kostnad i förhållande till den mängd genetisk information som analyseras. Begränsningarna med CES inkluderar avsaknaden av 100 % känslighet, oförmågan att upptäcka vissa typer av genetiska förändringar (t.ex. deletioner, duplikationer, trinukleotidrepetitioner, djupa introniska mutationer eller metyleringsdefekter) och det faktum att en diagnos kanske inte ger ytterligare information om sjukdomen eller förändrar hanteringen.
I rapporteringen av CES kan oavsiktligt upptäckta patogena varianter i gener som är associerade med välkända genetiska tillstånd rapporteras som sekundära resultat (78). Denna förteckning över rekommenderade sjukdomar har upprättats av American College of Medical Genetics (ACMG). GINA-lagen (Genetic Information Nondiscrimination Act) är ett viktigt övervägande när man beslutar om man ska välja om man vill ta del av eller inte ta del av information om tillfälliga fynd (79). GINA skyddar individer från missbruk av genetisk information vid sjukförsäkring och anställning, men inte vid livförsäkring. GINA skyddar följande genetiska information: medicinsk familjehistoria, bärartestning, prenatal genetisk testning, känslighetstestning och prediktiv testning samt analys av tumörer eller andra bedömningar av gen-, mutations- eller kromosomförändringar.
Hantering
Hanteringen av CDG beror till stor del på individens specifika symtom. Återkommande symtom hos patienter med CDG är bl.a. utebliven tillväxt, global utvecklingsförsening, kräkningar, stroke-liknande episoder och skelettmissbildningar. Klinisk eller subklinisk koagulopati, endokrinopati, hepatopati och hjärtfel ses också ofta. Grundläggande laboratorietester för att fastställa sjukdomens omfattning och rutinmässig övervakning rekommenderas, särskilt för PMM2-CDG. Dessa inkluderar leverfunktionstester, serumalbumin, sköldkörtelfunktionstester inklusive fritt T4, protein C, protein S, antitrombin III, faktor IX, urinanalys samt serumgonadotropiner och tillväxthormon.
Rekommenderad bilddiagnostik inkluderar ekokardiogram, ultraljud på njurarna, benålder, oftalmologisk undersökning för utvärdering av linsen, näthinnan, okulär rörlighet och intraokulärt tryck. Om inget annat anges rekommenderas rutinvaccinationer för vuxna och barn som drabbats av CDG. Antikroppstitrar bör erhållas efter vaccination eftersom patienterna kan ha ett suboptimalt immunogent svar. Profylaktisk påfyllning av koagulationsfaktorer före eventuella kirurgiska ingrepp kan vara nödvändig om det finns brister vid baslinjen.
Klinisk genetisk utvärdering bör genomföras för att diskutera de ärftliga aspekterna av CDG, samt för att upprätta ett medicinskt hem för dessa komplexa patienter. Det medicinska hemmet är vanligtvis den biokemiska genetiska avdelningen, även om genetik-, neuroutvecklings- eller neurologiavdelningar också har fungerat i denna egenskap om en särskild biokemisk genetisk avdelning inte är tillgänglig. Det är ofta nödvändigt att remittera specialistläkare till gastroenterologi, hematologi, endokrinologi, nutritionsstöd, logopedi, arbetsterapi, sjukgymnastik och matningsterapi, ortopedi och rehabiliteringsmedicin.
Målinriktade terapier och prognos
Behandlingen av majoriteten av CDG-typerna är i stort sett stödjande, med några få undantag. MPI-CDG är den mest effektivt behandlingsbara av alla CDG. Oral mannos omvandlas till mannos-6-fosfat av intracellulära hexokinaser, vilket innebär att enzymblocket kringgås och att det bristfälliga substratet produceras. Mannostillskott börjar vanligtvis med 1 g/kg kroppsvikt per dag, fördelat på 4-6 doser per dag. Medan den potentiellt livshotande proteinförstörande enteropatien är särskilt känslig för mannosbehandling kan leversjukdomen hos MPI-CDG fortsätta att utvecklas. Kliniska symtom förbättras snabbt och transferrin-CDT normaliseras under månader, även om leversjukdomen kan fortsätta att fortskrida under behandlingen (45,80,81).
Försiktighet bör iakttas vid tillskott av mannos under dräktighet, eftersom mannosadministrering i dräktiga hypomorfa fosfomannosisomeras-musmodeller resulterade i embryonal dödlighet och blindhet hos deras ungar (82). Dessutom har intravenös mannos förknippats med nedsatt medvetande och kramper, som försvann med glukosadministration (83).
Behandlingen av PMM2-CDG är till stor del stödjande och baserad på symtomatologi. Kommande kliniska prövningar om mannos-1-fosfat-substratersättningsbehandling är dock för närvarande under utveckling.
För andra CDG har olika orala enkla sockerarter undersökts i syfte att teoretiskt förbättra hypoglykosyleringen. Fukos har prövats för SLC35C1-CDG och galaktos för PGM1-CDG och SLC35A2-CDG med blandade resultat (84). D-galaktos på 1,0-2,5 g/kg/dag (max 50 gram) har visat sig förbättra hypoglykemi, koagulopati och endokrinopati hos PGM1-CDG (85,86). Galaktos har också visat sig förbättra endokrinopati och koagulopati hos TMEM165-CDG (87) och SLC39A8-CDG. Betydande klinisk förbättring rapporterades också hos SLC39A8-CDG-patienter som fick 15-20 mg/kg/dag MnSO4 (88). Kliniska försök pågår för att undersöka nyttan av N-acetylmannosamin (ManNAc) i GNE-CDG (89), och flera prekliniska försök pågår för andra CDG (90).
Trots medicinska framsteg finns det en betydande dödlighet för barn med CDG inom det första levnadsåret på grund av multiorgansvikt eller allvarlig infektion (91). Spädbarn med CDG kan visa sig med fulminant multiorgansjukdom, svårstoppade kramper eller svår hypoalbuminemi som utvecklas till anasarka. Vissa patienter svarar på aggressiv diures och albuminersättning medan andra är refraktiva till behandling. Natriumbutyrat har visat sig förbättra anfallskontrollen hos CAD-CDG och PIGM-CDG (92). Ketogen kost har också visat sig minska anfallsfrekvensen i vissa fall av PIGA-CDG (93). Under stroke-liknande episoder kan intravenös hydrering och upprätthållande av normalt blodglukos vara till hjälp medan underliggande vaskulär trombotisk eller blödande etiologi utesluts.
Med tillkomsten av tekniker för genomredigering och bättre förståelse av sjukdomsmekanismerna som omfattas av det diagnostiska paraplyet CDG, är framtiden för målinriktad terapeutisk utveckling fortfarande lovande.
Acknowledgements
Vi vill tacka Lynne Wolfe, ARNP och Donna Krasnewich, MD, PhD för att de försåg oss med kliniska foton som erhållits som en del av Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789). Vi vill också tacka Jenny Thies, MS, LGC för hennes expertis inom genetisk rådgivning.
Finansiering: IJ Chang stöds av National Institutes of Health T32GM007454.
Fotnot
Intressekonflikter: Författarna har inga intressekonflikter att deklarera.
Informerat samtycke: Författarna har inga intressekonflikter att deklarera:
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutationer i PMM2 som orsakar medfödda glykosyleringsstörningar, typ Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. Prevalensen av PMM2-CDG i Estland baserat på befolkningsbärarfrekvenser och diagnostiserade patienter. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Avsaknad av Hardy-Weinberg-jämvikt för den vanligaste PMM2-mutationen i CDG-Ia (congenital disorders of glycosylation type Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialinsyrabrist i serum och transferrin i cerebrospinalvätska i ett nyligen erkänt genetiskt syndrom. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG-nomenklatur: dags för en förändring! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Vad är nytt inom CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Medfödda störningar av glykosylering. Del I. Defekter i proteinets N-glykosylering. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellulära funktioner hos N-länkade glykaner. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Kliniska, laboratorie- och molekylära fynd och långtidsuppföljningsdata hos 96 franska patienter med PMM2-CDG (fosfomannomutas 2-kongenital störning av glykosylering) samt genomgång av litteraturen. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. En landsomfattande undersökning av PMM2-CDG i Italien: hög frekvens av en mild neurologisk variant associerad med L32R-mutationen. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG i Sydafrika: Genotyp-fenotypbeskrivning av fem nya patienter. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Olika glykosyltransferaser bearbetas differentiellt för sekretion, dimerisering och autoglykosylering. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In: Mora-Montes H. Redaktör. Glycans: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge:
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Hudmanifestationer vid CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF-systemet hos barn med medfödda glykosyleringsstörningar. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. Ett brett spektrum av kliniska presentationer vid medfödda glykosyleringsstörningar I: en serie på 26 fall. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertal tillväxt vid medfödd glykosyleringsstörning typ Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Kliniska och biokemiska egenskaper hos medfödd störning av glykosylering typ Ic, den första erkända endoplasmatiska retikulumdefekten i N-glykansyntesen. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1 – en nyligen upptäckt ärftlig metabolisk sjukdom. Multipla organmanifestationer, incidens 1/80 000, svårbehandlad. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Svår övergående myokardiell ischemi orsakad av hypertrofisk kardiomyopati hos en patient med medfödd glykosyleringsstörning typ Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Konotrunala hjärtfel hos tre patienter med kongenital glykosyleringsstörning typ Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Perikardiell och abdominal vätskeansamling vid medfödd glykosyleringsstörning typ Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Kliniska särdrag hos vuxna med medfödda glykosyleringsstörningar av typ Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Kolhydratbristande glykoproteinsyndrom. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Kliniska och molekylära egenskaper hos medfödd störning av glykosylering hos patienter med typ 1 sialotransferrinmönster och olika etniskt ursprung. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc-modifiering av pälsproteinet hos Potyvirus Plum pox virus ökar virusinfektionen. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Skandinaviska CDG-Ia-patienter: genotyp-/fenotypkorrelation och grundmutationernas geografiska ursprung. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 franska vuxna patienter med PMM2-kongenital glykosyleringsstörning: resultat av den klassiska pediatriska fenotypen och beskrivning av en sent insatt fenotyp. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinala egenskaper hos den medfödda glykosyleringsstörningen PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): fenotypiskt spektrum av genotypen R141H/F119L. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Sköldkörtelfunktion vid PMM2-CDG: diagnostiskt tillvägagångssätt och föreslagen behandling. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental utveckling hos en patient med medfödd glykosyleringsstörning (CDG) typ Ia med multisystemisk inblandning (intermediär fenotyp). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Nya störningar i kolhydratmetabolismen: medfödda glykosyleringsstörningar och deras inverkan på det endokrina systemet. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogenitet i den kliniska presentationen från multivisceralt misslyckande till hyperinsulinaemisk hypoglykemi som ledande symtom hos tre spädbarn med fosfomannomutasbrist. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endokrinologi vid kolhydratbristande glykoproteinsyndrom typ 1 från födsel till ungdom. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadal funktion och glykoproteinhormoner vid kolhydratbristande glykoproteinsyndrom (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. Det kliniska spektrumet av fosfomannosisomerasbrist, med en utvärdering av mannosbehandling för CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Hjärtkomplikationer till följd av medfödda glykosyleringsstörningar (CDG): en systematisk genomgång av litteraturen. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Kolhydratbristande glykoproteinsyndrom typ Ib. Brist på fosfomannosisomeras och mannosbehandling. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Svår hypoglykemi som ett symtom på kolhydratbristande glykoproteinsyndrom. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemisk hypoglykemi som ett tecken på fosfomannoseisomerasbrist: En ny manifestation av kolhydratbristande glykoproteinsyndrom som kan behandlas med mannos. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Kliniskt användbart genkort för: ALG6 defekt medfödd glykosyleringsstörning. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Luftvägsslem: Från produktion till sekretion. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparansulfat i skelettets utveckling, tillväxt och patologi: fallet med ärftliga multipla exostoser. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Diverse icke-inflammatoriska muskuloskeletala tillstånd. Hyperfosfatematisk familjär tumörkalcinos (FGF23, GALNT3 och αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Ablation av Galnt3-genen leder till låga cirkulerande intakta koncentrationer av fibroblasttillväxtfaktor 23 (Fgf23) och hyperfosfatemi trots ökat Fgf23-uttryck. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. En mutation i ett gangliosidbiosyntetiskt enzym, ST3GAL5, resulterar i salt &peppar-syndromet, en neurokutan sjukdom med förändrad glykolipid- och glykoproteinglykosylering. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutationer i B4GALNT1 (GM2-syntas) ligger bakom en ny störning av gangliosidbiosyntesen. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA-mutationer orsakar epileptiska encefalopatier med tidig debut och distinkta drag. Neurologi. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorfa mutationer i PGAP2, som kodar för ett GPI-ankor-remodellerande protein, orsakar autosomalt recessivt intellektuellt funktionshinder. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. Fenotypen av en germlinemutation i PIGA: genen somatiskt muterad i paroxysmal nattlig hemoglobinuri. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutationer i PIGO, en medlem av GPI-ankersyntesvägen, orsakar hyperfosfatasi med mental retardation. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. Ett nytt syndrom med intellektuell funktionsnedsättning orsakat av GPI-ankarbrist på grund av homozygota mutationer i PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutationer i glykosylfosfosphatidylinositolgenen PIGL orsakar CHIME-syndromet. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. Det genotypiska och fenotypiska spektrumet av PIGA-brist. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nattlig hemoglobinuri (PNH) orsakas av somatiska mutationer i PIG-A-genen. EMBO J 1994;13:110-7.
- Brodsky RA. Paroxysmal nattlig hemoglobinuri. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. Masspektrometriens inverkan på diagnosen av medfödda glykosyleringsstörningar. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessiv dilaterad kardiomyopati på grund av DOLK-mutationer beror på onormal O-mannosylering av dystroglykan. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. Kostnad och diagnostiskt utbyte av exomsekvensering för barn med misstänkta genetiska sjukdomar: en benchmarkingstudie. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Strömningar i samtida etik. GINA, ADA och genetisk diskriminering i arbetslivet. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Återkommande tromboembolism hos ett barn med en medfödd glykosyleringsstörning (CDG) typ Ib och behandling med mannos. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Utveckling av leversjukdom trots mannosebehandling hos två patienter med CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannostillskott framkallar embryonal dödlighet och blindhet hos hypomorfa möss med fosfomannosisomeras. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Kramper och stupor under intravenös mannoseterapi hos en patient med CDG-syndrom typ 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Fukostillskott vid leukocytadhesionsbrist typ II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Ärftlig glykosylfosfatidylinositolbrist: en behandlingsbar CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Oralt D-galaktostillskott i PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Galaktostillskott hos patienter med TMEM165-CDG räddar glykosyleringsdefekterna. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8-brist: biokemisk korrigering och stor klinisk förbättring genom manganbehandling. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Orala monosackaridterapier för att vända njur- och muskelhyposialylering i en musmodell av GNE-myopati. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: Från bänk till sängkant. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Human glykosyleringsstörningar. Marino PA, redaktör. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogen kost – en ny behandling för tidig epileptisk encefalopati på grund av PIGA-brist. Brain Dev 2016;38:848-51.