- 概要
- 疫学
- 生化学的分類と命名法
- Genetics
- 病態生理
- N-linked protein glycosylation defects
- O-結合型糖鎖形成不全とN-およびO-結合型糖鎖形成不全
- 脂質糖鎖形成とGPIアンカー生合成の異常
- 臨床症状
- N-linked protein glycosylation defects
- PMM2-CDG (CDG-Ia, PMM2 deficiency)
- MPI-CDG (CDG-Ib, mannosephosphate isomerase deficiency)
- ALG6-CDG (glucosyltransferase 1 deficiency)
- O-linked glycosylation defects and combined N- and O-linked glycosylation defects
- 脂質糖鎖およびGPIアンカー生合成異常
- 診断
- N-linked protein glycosylation defects
- O-linked glycosylation defects and combined N- and O-linked glycosylation defects
- 脂質糖鎖形成およびGPIアンカー生合成不全
- 分子解析
- 標的治療と予後
- 謝辞
- 脚注
概要
グリコシレーションは、さまざまな細胞内経路でタンパク質や脂質に糖残基を付加する過程である。 先天性糖鎖異常症(CDG)は、糖鎖合成または修飾経路に沿った様々なステップの欠陥によって引き起こされる、遺伝的にも臨床的にも不均質な100以上の疾患のグループである。
CDGは一般に、発達遅延、成長障害、筋緊張低下、神経障害、肝障害、凝固障害などの多臓器症状を呈する。 また、眼、皮膚、心疾患、顔面異形成を伴うこともある。 神経学的変化や認知機能の遅れは大部分の患児に見られますが、神経学的症状を示さない症例やタイプさえあります。
血清糖鎖欠損トランスフェリン(CDT)分析は、CDGが疑われる患者における第一選択のスクリーニング検査であるが、シアル酸欠損を伴うN-グリコシル化欠損の検出に限定されている。 次の検査としては、ドリコール結合糖鎖分析および遺伝子検査がある。 CDGの中には治療可能なものもあるので、この指数関数的に増加する疾患群の早期診断が重要である。 糖鎖異常症の治療は主に支持療法であるが、MPI-CDG、SLC35C1-CDG、PIGM-CDG、PGM1-CDGに対しては標的治療が可能である。 これらの治療法の詳細は、後述の「標的治療と予後」の項に記載した。
疫学
すべてのタイプのCDGの総体的な発生率と有病率は十分に確立されていないが、世界中でほとんどすべての民族的背景から患者が報告されており、両性は等しく罹患している。 53の遺伝子における既知の病原性バリアントのキャリア頻度から、ヨーロッパおよびアフリカ系アメリカ人集団における推定有病率は1/10,000である(1-4)。 最も多く診断されているCDGであるPMM2-CDGの有病率は、孤立した報告に基づいて、オランダの集団では1/20,000から、エストニアでは1/77,000である(5,6)。
生化学的分類と命名法
現在、CDGは大きく分けて、(I)N-結合型糖鎖形成、(II)O-結合型糖鎖形成、(III) N-, O-結合/多重糖鎖形成複合、および (IV)lipid, glycosylphosphatidylinositol (GPI) anchor biosynthesis defects に分類され、その分類は、N-結合型糖鎖の形成が、O-結合型と多重糖鎖の複合、および脂質とGPIの合成の欠陥に分類されている。
以前はCDG type Iaとして知られていたN-結合型タンパク質グリコシル化異常PMM2-CDGは、1980年にJaekenによって報告された最初のCDGで、現在でも圧倒的に多いCDGである(7)。 PMM2-CDGは、当初、患児の血清トランスフェリンの等電点電気泳動で見られる複数の血清糖蛋白質異常から「糖質欠乏性糖蛋白質症候群」と命名されたが、現在では、PMM2-CDGは「糖質欠乏性糖蛋白質症候群」と呼ばれている。 歴史的には、CDGはトランスフェリンアイソフォーム解析のパターンによって分類されていた。タイプIのパターンは、細胞質または小胞体に局在するドリコール結合型糖鎖の組み立ておよび転移障害に起因し、タイプIIのパターンはゴルジ体におけるプロセッシングの障害に起因していた。 この分岐点から、CDGは発見順にアルファベット順に命名された。
分子診断の普及に伴い、CDGの命名法は2008年に疾患の分子的病因を特定するために更新されたが、これは以前に確立した二元的カテゴリーにうまく当てはまらない経路や疾患の急激な増大を反映したものである。 現在、CDGの命名法は、影響を受ける遺伝子名(非イタリック、遺伝子名はwww.genenames.org)の後に-CDG(例:PMM2-CDG)を付けて表記されている(8)。
Genetics
先天性の糖鎖障害の大部分は、常染色体劣性遺伝し、無症状の(キャリア)親から1つづつ変異が受け継がれます。 遺伝子診断の確立には、通常、次世代シーケンサーを用いた分子生物学的検査が必要である。 既知の変異体に関する両親の検査により、遺伝かde novo発生かを確認することができます。 常染色体劣性遺伝の場合、罹患者の兄弟姉妹および各妊娠における再発リスクは、罹患者が25%、無症候性保因者が50%、非罹患者が25%である。
一握りのCDGは常染色体優性遺伝(Nリンク性。 GANAB-CDG、PRKCSH-CDG、O-linked: Ext1/Ext2-CDG、Pofut1-CDG、Poglut1-CDG)。 X連鎖は少ない(ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG)。 CDGのほとんどの優性型および一部のX連鎖型は、de novo変異によるものです。 具体的な疾患や遺伝子については、後述の「病態生理」の項で説明する。
CDGのすべての公表遺伝子の変異データは、Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php)で入手可能である。 特定の遺伝子変異に関する情報は、Leiden Open Variation Database with integrated in silico pathogenicity tools (http://www.lovd.nl/3.0/home)で入手可能である。 特定の遺伝子に関する臨床的な概要はOnline Mendelian Inheritance in Man (http://www.omim.org/) やより限定的な範囲ではGeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/) で見ることができる。
病態生理
CDGは現在までに130種類以上報告されている(9,10)。 このように、CDGは糖鎖修飾経路が普遍的に存在するため、その生化学的な病態は極めて多様である。 多くのタンパク質や脂質(スフィンゴ脂質や糖脂質)が、様々な細胞内で単糖やオリゴ糖のグリコシル化(総称して糖鎖)を受けており、このような糖鎖を持つタンパク質や脂質がCDGとなる。 細胞内におけるその位置は多様であるが、多くの欠陥は小胞体あるいはゴルジ装置内で生じる。
タンパク質のうち、糖鎖はポリペプチド鎖に結合しており、N-糖鎖はアスパラギン(Asn)のアミド基に結合し、O-糖鎖はセリンまたはスレオニンの水酸基に結合している。 N型糖鎖の合成には、細胞質でヌクレオチドが結合した糖鎖が構築され、小胞体で組み立てられ、ゴルジ体で処理されるという段階的な工程が必要である。 一方、O-グリカン合成は、組み立ては必要だがプロセシングは必要ないため、O-グリコシル化の欠陥はゴルジ装置で主に起こる。
N-linked protein glycosylation defects
N-グリコシル化は、Asn-X-Ser/Thrアクセプターサイト内のAsn残基の側鎖アミド基に糖鎖構造が共有結合し、小胞体に転送されて再構築され、ゴルジ体でさらにN-グリカン鎖が改変される (11,12).
PMM2-CDG はホスホマンノムターゼ2 (PMM2) 遺伝子の変異により発症し、グアノシン二リン酸 (GDP) によるマンノース合成の第2段階でマンノース6リン酸をマンノース1リン酸に細胞質変換する酵素の欠損が起こる。 ほとんどの患者は複合ヘテロ接合性の病原性ミスセンス変異を保有している(www.lovd.nl/PMM2)。 最も一般的な再発性の病原性変異体p.Arg141Hisは、ヨーロッパ系の罹患者の約40%に認められ、p.Phe119Leuも北ヨーロッパで頻繁に認められる(1)。 PMM2-CDGでは遺伝子型と表現型の相関が報告されている(3,13,14)。
MPI-CDG はマンノースリン酸アイソメラーゼ(MPI)遺伝子の病因変異によるホスホマンノースイソメラーゼ(MPI)欠乏症で常染色体劣性遺伝であり、その原因としてマンノースリン酸アイソメラーゼ(MPI)遺伝子に変異があり、その結果マンノースリン酸が欠損することが知られている。 MPIは通常、GDP-マンノース合成の第一段階(フルクトース-6-リン酸からマンノース-6-リン酸への変換)を触媒するが、フルクトース-6-リン酸は解糖系経路でも代謝されるため細胞内には蓄積されない。 したがって、生化学的にはPMM2-CDGと類似していますが、MPI-CDGはそれほど重大な神経学的および多系統の病変を引き起こすことはありません。 CDTは、MPI-CDGのスクリーニング検査としても選択され、1型のパターンを示します。 ALG6-CDGはALG6の変異によって起こる劣性疾患で、3つのグルコース分子がドリコール結合マンノース中間体に異常付着し、下流で血清糖タンパクの低グリコシル化を引き起こす(15)。
O-結合型糖鎖形成不全とN-およびO-結合型糖鎖形成不全
O-グリコシル化は、ゴルジ装置内の糖転移酵素によってタンパク質のセリン、スレオニン、ヒドロキシリシン残基に糖鎖が段階的に付加されている (16). 5949>
脂質糖鎖形成とGPIアンカー生合成の異常
GPIアンカーは小胞体で順次組み立てられ、ゴルジ体内で修飾される糖脂質である。 酵素欠損によるGPIアンカー生合成障害は、発見順のアルファベット順で命名されており、組み立ての段階を時系列で表しているわけではない。 GPIアンカーは一度合成されると、細胞膜上に存在し、何百もの細胞表面タンパク質と結合し、多くの細胞機能を発揮する。
臨床症状
糖鎖形成経路がいたるところに存在することから、CDGはほぼすべての臓器が関与していると考えられるが、ほとんどの症例で神経系に異常が見られる。 MPDU1-CDG、DOLK-CDG、SRD5A3-CDG(図1)、PIGL-CDGなどの魚鱗癬を呈するCDGもある(18,19)。 ほぼ全てのCDGは生後数年以内に多臓器疾患を呈するが、一部は単一の臓器系のみを侵す(例. 網膜はDHDDS-CDG、神経筋接合部はALG2-CDG、ALG14-CDG、CFPT1-CDG、脳はST3GAL3-CDG、TUSC3-CDG、皮膚や骨格筋はPOGLUT1-CDG、POFUT1-CDG、軟骨はEXT1/EXT2-CDG、肝臓はTMEM199-CDG、赤血球はSEC23B-CDG) などです。 発症年齢と重症度は、新生児致死からほぼ無症状の成人まで、またその中間の順列になることもある。 最もよく報告される症状は、発達遅延、成長障害、筋緊張低下、神経学的異常、低血糖、および肝臓、眼、皮膚、消化管、免疫学的、骨格、凝固異常の多様性である(19)。
多くのCDG亜型の完全な表現型は、報告例が稀なため、まだ完全に解明されていない。 したがって、CDGは、多臓器疾患のどのような状況においても、特に神経学的要素を有する症例や原因不明の非特異的な発達遅滞において考慮すべきである。
多数の症状の病態生理学はまだ解明されていないが、特定の糖鎖形成経路と特定の臨床症状の関係が明らかにされつつある。 例えば、多くのCDGで見られるfailure to thriveは、IGF-1、ALS、IGFBP-3などのインスリン成長経路にあるいくつかの糖タンパク質の低グリコシレーションと形成障害に起因している(20)。 この複雑な疾患群に対する理解が深まるにつれ、CDGは診断のつかない患者においてますます認識されるようになってきている。 異なるCDGの臓器系への関与はTable S1にまとめられている。 5949>
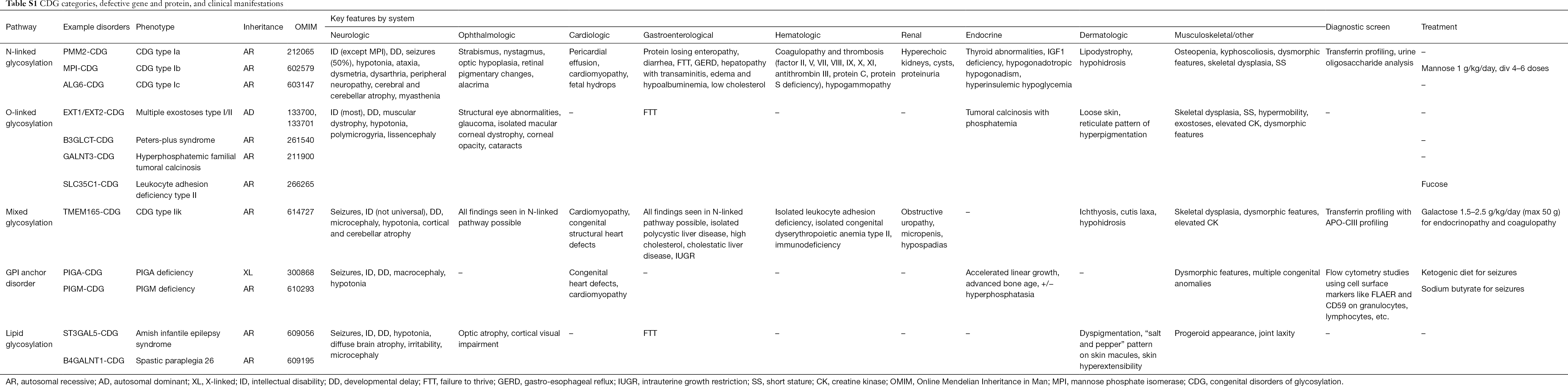
Full table
N-linked protein glycosylation defects
CDG として最も多く診断されている N-linked glycosylation disorder の表現型が、しばしば典型例と謳われることがある。
PMM2-CDG (CDG-Ia, PMM2 deficiency)
PMM2-CDG は最も一般的なCDGであり、世界で700例以上報告されている。 乳児期の多系統の重症化、小児期の神経疾患や発達遅延、成人期の安定した知的障害を特徴とする(21,22)。
乳児期のPMM2-CDGは、出生直後に斜視や眼球運動異常、小脳低形成、低血圧、精神運動遅延、運動失調、低血圧、反射減退という神経系異常で代表される症状が現れる。 また、肝疾患、ネフローゼ症候群、腎嚢胞、心嚢液貯留、肥大型心筋症、発育不全、多臓器不全を呈し、最大で20%の患者が生後1年以内に死亡する(21、23-28)
PMM2-CDG患者では異形性の特徴群が報告されている(図2、3)。 これらの特徴には、低形成小脳、顔面異形(すなわち、大きな異形成耳)、逆さ乳首、臀部または恥骨上部の脂肪組織の異常分布が含まれ、これらは年齢とともに消失することがある(14、21、29~32歳)。 患者は、外向的で幸せな態度をとることが報告されている。 症状は非常に多様であるが、患児の70%以上に斜視が認められる(21,23,33-35)。 反転した乳頭や異常な脂肪パッドは約25-50%の患者に見られる(36)。

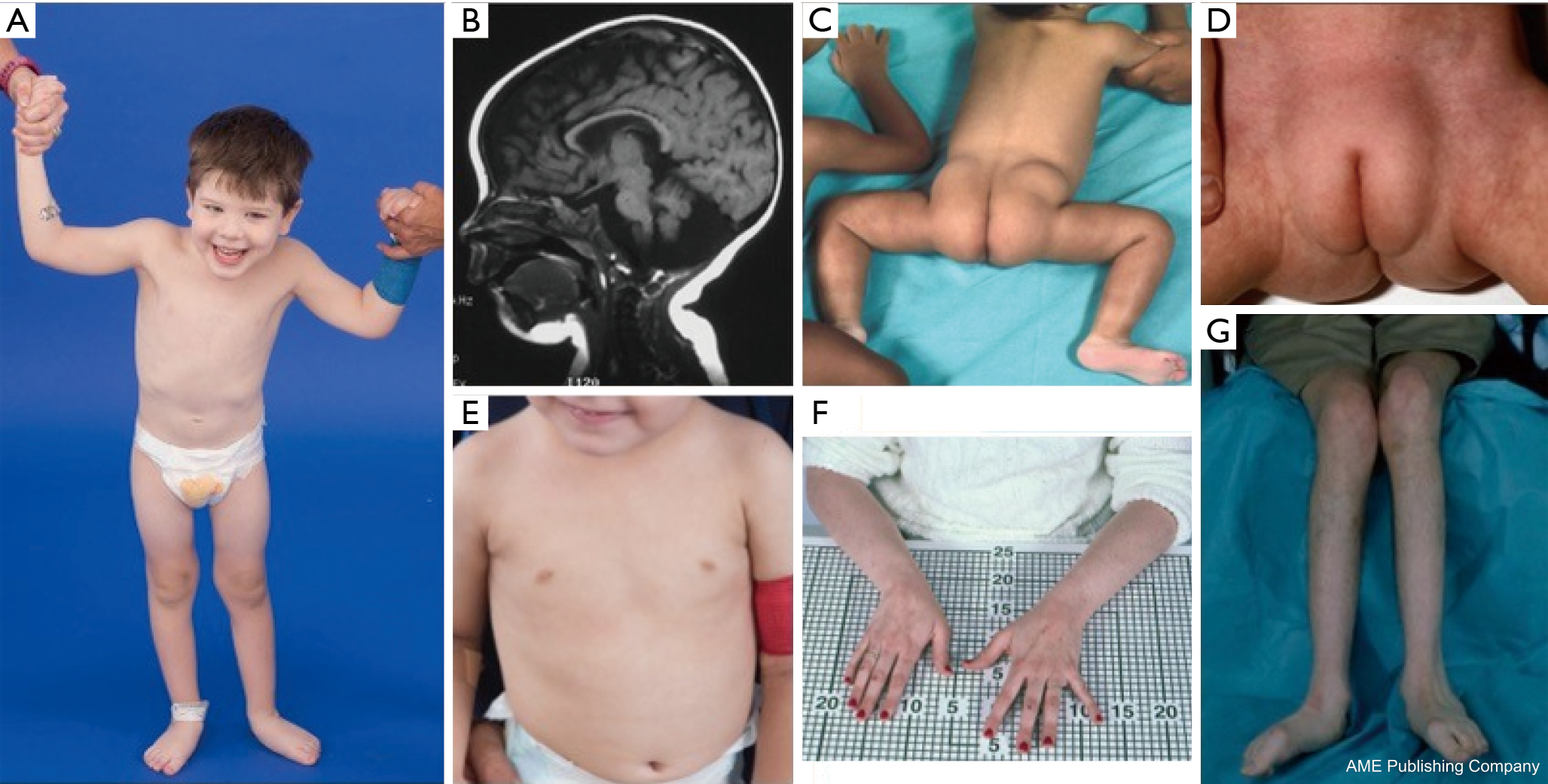
小児期には、患児は網膜色素変性症、脳卒中様のエピソードおよび発作、言語および運動の遅れ、および末梢神経障害を発症することがあります。 体質的には、摂食・消化器系の異常による成長障害、および全体的な発達遅滞が一般的である。 臨床的影響を伴わない肝トランスアミナーゼの上昇が観察されることがあるが、一般的に5歳までに正常化し、病気によって時々変動する(21,24)。 肝線維化が疑われない限り、CDGで肝生検が適応されることは稀である(1)。 臨床的な甲状腺機能低下症はまれであるが、CDG患者は甲状腺ホルモンとフリーT4を測定する必要があり、甲状腺結合グロブリン(TBG)の低下と甲状腺刺激ホルモン(TSH)の一過性の上昇を示すことがある(37)。 PMM2-CDGでは肝・胆管奇形は報告されていない。
成人のPMM2-CDGでは、安定した認知遅延、末梢神経障害、骨減少症や骨粗鬆症を伴う進行性の胸部・脊椎後彎症を伴い、7、8十代まで生きることもある(34)。 小脳失調は、多系統の病変とともに、ますます認識される症状である(38-40)。 高プロラクチン血症、高血糖を伴う成長ホルモン分泌、インスリン抵抗性、高インスリン血性低血糖などの内分泌異常がある(41,42)。 罹患女性では、低ゴナドトロピン性性腺機能低下症により、二次性徴の欠如や卵巣の欠落が生じることがあります(41,43,44)。
MPI-CDG (CDG-Ib, mannosephosphate isomerase deficiency)
MPI-CDG の特徴は、神経障害がほとんどなく、いくつかの症状はマンノースの経口投与で治療が可能であることである(2)。 症状は主に肝腸系で、異形症状や認知機能の遅れはない。 患者は典型的には、再発性嘔吐、著しい低血糖、成長不良、生命を脅かす可能性のある蛋白質廃棄腸症、肝臓の線維化変化、胆管拡張を呈する(45-51)。
ALG6-CDG (glucosyltransferase 1 deficiency)
ALG6-CDG は2番目に多いN-グリコシル化異常で、PMM2-CDGと似ているが表現型が穏やかであることが特徴である。 ALG6-CDGの患者は、成長不良、発達遅延、筋緊張低下、発作、斜視、運動失調、凝固障害、顔面異形(すなわち、低位耳、多毛、巨舌症)などがある。 MPI-CDGと同様に、蛋白喪失性腸症が見られることもあります。 さらに、罹患者は腕足らずや指の奇形、側弯などの骨格異常を有することがある。 網膜色素変性症や小脳低形成は典型的な例ではありません(52)。
O-linked glycosylation defects and combined N- and O-linked glycosylation defects
GAG (glycosaminoglycans) と上皮表面を含むムチン含有タンパク質におけるO-グリカンの存在により、GAG合成の異常は典型的には骨格形成異常や結合組織疾患を引き起こす。 患者は、神経症状に加え、筋骨格系、皮膚、関節の異常(例えば、関節弛緩症、多発性外骨腫、軟骨・骨肉腫)を呈することがある(54-56)。 例えば、N-アセチルガラクトサミニルトランスフェラーゼ3(GALNT3)は、リン酸化ホルモンであるFGF23をO-グリコシル化し、タンパク質分解による切断を防ぎ、そのまま分泌できるようにする働きがある。 GALNT3欠損は高リン血症と異所性石灰化を特徴とする家族性腫瘍性石灰化症につながる(57,58)。
脂質糖鎖およびGPIアンカー生合成異常
糖脂質およびそのシアリル化誘導体のガングリオシドは主に神経細胞に発現している。 ガングリオシドの分解に欠陥があると蓄積し、よく知られたライソゾーム貯蔵病となる。 一方、ST3GAL5-CDGやB4GALNT1-CDGのようなガングリオシドの生合成の欠陥は極めて稀であり、重篤な神経変性疾患を引き起こす。 GPIアンカー生合成経路の多くの遺伝子に変異があると、様々な先天異常、知的障害、てんかんが引き起こされる(59,60)。 最も特徴的なGPI生合成異常はX連鎖性PIGA欠損症であり、低血圧、多発性脳異常、顔面異形症を伴う小児けいれんを呈する。 また、皮膚、肝臓、心臓、腎臓の疾患も多様である(61-69)。 PIGAの一部の変異は、表現型的に異なる疾患である発作性夜間ヘモグロビン尿症(PNH)、骨髄不全の後天性疾患(70,71)を引き起こす。
診断
CDGが臨床的に疑われた場合、最初のステップはCDTとN-グリカン試験を含む血漿または血清中の生化学CDG試験をオーダーすることである。 血清CDTおよびN-グリカン分析はN-グリコシル化欠損のみを検出できるため、孤立したO-グリコシル化欠損またはGPIアンカー欠損の鑑別には有用ではないだろう。 トランスフェリンのアイソフォーム解析は、もともとトランスフェリンの等電点集束によって行われていた。N-グリカン合成の失敗によってシアル酸が部分的に欠乏し、これが血清トランスフェリンの電荷を変え、その結果電気泳動場での陰極の移動を変化させるからである。 しかし、質量分析に基づくトランスフェリンとN-グリカンの分析は、質量と電荷によるオリゴ糖の特異的変化を同定することにより、現在では等電点集束にほぼ取って代わられている(72)。
N-linked protein glycosylation defects
Serum transferrin CDT results are reported as the ratio of mono-oligosaccharide/di-oligosaccharide transferrin.これは、血清トランスフェリンのCDTの結果が、単糖/二糖の割合で報告されている。 a-オリゴ糖/ジ・オリゴ糖トランスフェリン、トリシアロ/ジ・オリゴ糖トランスフェリン、アポリポ蛋白CIII-1/アポリポ蛋白CIII-2、アポリポ蛋白CIII-0/アポリポ蛋白CIII-2の比率を示す。 これらの定量的な結果は、所見のパターンの解釈も伴ってくる。
タイプIパターンのトランスフェリンCDTは、ジおよびアシアロトランスフェリンのバンドの増加を特徴とし、細胞質または小胞体におけるN-糖鎖合成の欠損を示す。
I型の血清トランスフェリンCDTパターンが検出された場合、PMM2欠損症またはMPI欠損症は鑑別の最前線に立つべきである。なぜならPMM2-CDGは最もよく見られるCDGであり、MPI-CDGは治療可能だが放置すると致命的となる可能性があるからだ。 診断を区別するために、N-グリカンプロファイリング、分子配列決定、または酵素試験を実施する必要がある。 PMM2-CDG または MPI-CDG の診断は、PMM2 または MPI の二遺伝子変異を示す分子検査、および遺伝子変異の病原性が不明な場合は白血球または線維芽細胞における PMM または MPI 酵素活性によって確定される。 ALG-CDGの大部分はN-グリカン分析あるいは分子解析によりPMM2あるいはMPI-CDGと区別できる(15)。
II型血清トランスフェリンCDTパターンはN-アセチルグルコサミニルトランスフェラーゼ(GnT)II欠損などのGolgi欠損(CDG type IIA、MGAT2-CDG)を示している。 アポリポ蛋白CIII(Apo-CIII)アイソフォーム分析は、ゴルジ体におけるムチン型O-グリコシル化欠損を測定するため、II型CDTプロファイルの補完的検査となる。 CDTやApo-CIIIによるII型CDGの検出感度は限られている。 したがって、N-グリカンおよびO-グリカンのプロファイリングと分子パネルまたはエクソームシーケンスは、これらの臨床検査が可能な場合に実施されるべきである。 トランスフェリンのグリコシレーションパターンは散発的に正常化することがある。 遺伝性果糖不耐症の急性クリーゼ、ガラクトース血症、急性肝疾患、いくつかの細菌感染症では、偽陽性が得られることがある。 生化学的なCDG検査はいずれもすべてのCDGをスクリーニングできないため、スクリーニング結果が正常であっても、臨床的に強く疑われる場合には、分子遺伝子パネル検査またはエクソームシーケンスを実施することがある。
O-linked glycosylation defects and combined N- and O-linked glycosylation defects
トランスフェリンアイソフォーム解析では単独のO-グリコシル化欠損を検出できないため、診断には分子配列の決定が不可欠である。 CDT、ApoCIII分析、血漿中のN-グリカンおよびO-グリカン分析により、N-およびO-結合型糖鎖形成不全が複合的に検出できる。
脂質糖鎖形成およびGPIアンカー生合成不全
血液顆粒球のフローサイトメーターでCD16およびCD24などのGPIアンカーの細胞表面発現を測定する。 PIGA遺伝子の後天的変異によるPNHの検査として、白血球または赤血球のフローサイトメトリーによる特定のGPIアンカー細胞表面タンパク質の分析が臨床的に可能である。 PNH検査では他のGPIアンカー欠損の異常が見つかることもあるが、診断はほとんどが分子解析に依存している。
分子解析
CDGの診断で最も収率が高いのは次世代ベースの遺伝子配列決定パネルまたはクリニカルエクソームシーケンス(CES)である。 遺伝子シークエンスは、遺伝子のヌクレオチド配列、または「文字」の綴りを校正し、遺伝子の機能に影響を与える変化があるかどうかを判断する。 ヒトのゲノムは300万個のヌクレオチドから構成されていますが、機能的なタンパク質産物に翻訳されるのは、エクソンと呼ばれるそのうちの1~2%にすぎません。 エクソンの間に散在する、翻訳されない残りの非コードDNAはイントロンと呼ばれる(74)。 CESは、ヒトゲノムに含まれる約2万個の遺伝子のうち、染色体中の遺伝物質の少数派を占めるが、病気の原因となる(病原性の)変異体を含む可能性が高い、ほぼすべての既知のエクソンを検査するものである。 CESはまた、ミトコンドリアDNA(mtDNA)配列決定も行うことができ、ミトコンドリア内に存在し、母系のみに遺伝する小さな、核外の、円形のDNAを調べる。
CESの考えられる結果には陽性、陰性、および意義不明の変異体がある。 陽性とは、疾患の原因となる既知の変異体(すなわち、病原性)が同定され、診断、自然歴、予後、再発リスク、治療オプションが検討されることである。 陰性結果は、検出可能な病原性バリアントが同定されなかったことを意味する。 意義不明の変異体(VUS)とは、遺伝的変化は確認されたものの、特定の遺伝的変化について、それが疾患の原因であるかどうかを明確に知るための情報が十分でないことを意味します。 各個人の DNA にはばらつきがあることが予想されるため、比較のために両親の検体を同時に検査す ることは、検査室および臨床における結果の解釈を助けることができる。 CES の診断率は 30-35%と推定され、遺伝子の発見とヒトゲノムに関する知識の進歩に伴い、時と共に上昇し ている(75-77)。 CESは、その迅速な処理と、解析された遺伝情報の量に対する相対的なコストの低さから、第一選択の広範な遺伝子検査として、ますます注文されるようになっている。 CESの限界としては、感度が100%ではないこと、ある種の遺伝子変化(例えば、欠失、重複、トリヌクレオチド反復、深いイントロン変異、メチル化異常)を検出できないこと、診断によって疾患に関する追加情報が得られない、あるいは管理が変わる可能性があること、などが挙げられる。
CESの報告では、有名な遺伝病と関連する遺伝子で偶然検出された病原性バリアントは、副所見として報告することができる(78)。 この推奨疾患リストは、米国医遺伝学会(ACMG)が監修したものである。 遺伝子情報非差別法(GINA)は、付随的所見の学習を選択するかしないかを決定する際に重要な考慮事項である(79)。 GINAは、健康保険や雇用における遺伝情報の悪用から個人を保護するが、生命保険には適用されない。 GINA は以下の遺伝情報を保護する:家族病歴、キャリア試験、出生前遺伝子検査、感受性及び予測試験、腫瘍の分析又は遺伝子、突然変異、染色体変化に関する他の評価。 CDG患者における再発性の症状には、成長不良、全体的な発達遅延、嘔吐、脳卒中様のエピソード、骨格異常が含まれる。 臨床的または亜臨床的な凝固障害、内分泌障害、肝障害、および心臓障害も一般的に認められます。 特に PMM2-CDG の場合、疾患の程度を把握するためのベースラインの臨床検査と定期的なモニタリングが推奨されます。 肝機能検査、血清アルブミン、遊離T4などの甲状腺機能検査、プロテインC、プロテインS、アンチトロンビンIII、第IX因子、尿検査、血清ゴナドトロピンおよび成長ホルモンなどです。
推奨する画像診断は、心エコー、腎エコー、骨年齢、眼科での水晶体、網膜、眼の動き、眼圧の評価です。 特に指定がない限り、CDGに罹患した成人および小児には、定期的な予防接種が推奨される。 患者は免疫原性反応が不十分である可能性があるため、ワクチン接種後に抗体価を測定する必要があります。
臨床遺伝学的評価は、CDGの遺伝的側面について議論し、これらの複雑な患者のためのメディカルホームを確立するために実施されなければならない。 メディカルホームは一般的に生化学遺伝学サービスであるが、生化学遺伝学専門サービスが利用できない場合は、遺伝学、神経発達、または神経学部門もこの役割を担っている。 消化器内科、血液内科、内分泌内科、栄養サポート、言語、作業、理学、摂食療法、整形外科、リハビリテーション科などの専門医の紹介がしばしば必要となる。
標的治療と予後
大部分のCDGタイプに対する治療は、いくつかの例外はあるが、ほぼ支持的なものである。 MPI-CDGは、すべてのCDGの中で最も効果的な治療法である。 経口マンノースは、細胞内のヘキソキナーゼによってマンノース-6-リン酸に変換されるため、酵素ブロックを回避して欠損基質を生成することができる。 マンノースの補給は通常、1日1g/kg体重から開始し、1日4~6回に分けて投与する。 生命を脅かす可能性のあるタンパク質消耗性腸症は、特にマンノース治療に反応するが、MPI-CDGの肝疾患は進行し続ける可能性がある。 臨床症状は急速に改善し、トランスフェリンCDTは数カ月で正常化するが、肝疾患は治療により進行し続けることがある(45,80,81)。
妊娠中のホスホマンノースイソメラーゼ低型マウスモデルへのマンノース投与により、仔に胚致死と失明が発生したので妊娠中の補給は注意が必要である(82)。 また、マンノースを静脈内投与すると意識障害や痙攣が起こるが、グルコース投与で消失する(83)。
他のCDGに対しては、理論的に低グリコシレーションを改善する目的で、様々な経口単糖が検討されている。 SLC35C1-CDGにはフコースが、PGM1-CDGとSLC35A2-CDGにはガラクトースが試みられたが、結果はまちまちであった(84)。 D-ガラクトースは1.0-2.5g/kg/day(最大50g)でPGM1-CDGの低血糖、凝固障害、内分泌障害を改善することが証明されている(85,86)。 ガラクトースは、TMEM165-CDG (87)とSLC39A8-CDGの内分泌障害と凝固障害を改善することも示されている。 SLC39A8-CDG患者において、15-20mg/kg/dayのMnSO4投与でかなりの臨床的改善が見られたことも報告されている(88)。 GNE-CDGにおけるN-アセチルマンノサミン(ManNAc)の有用性を調べる臨床試験が進行中であり(89)、他のCDGについてもいくつかの前臨床試験が進行中である(90)。
医学的進歩にもかかわらず、CDG患者の生後1年間の死亡率は多臓器不全または重度の感染によるものが大きい (91). CDGの乳児は、劇症の多臓器疾患、難治性の発作、または無アルカリ性に進行する重度の低アルブミン血症を呈することがある。 積極的な利尿とアルブミン補充に反応する患者がいる一方で、治療に難渋する患者もいる。 酪酸ナトリウムは、CAD-CDG と PIGM-CDG において発作のコントロールを改善することが証明されている (92)。 ケトジェニックダイエットは、PIGA-CDGのいくつかの症例で発作の頻度を減少させることが示されています(93)。 脳卒中様エピソードでは、血管血栓や出血の病因を除外しながら、静脈内水分補給と正常血糖の維持が有用である。
ゲノム編集技術の出現とCDGの診断の傘に含まれる疾患のメカニズムの理解により、標的治療開発の将来は有望であると考えられる。
謝辞
「Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation」(NCT02089789)の一環として得られた臨床写真を提供していただいたLynne Wolfe, ARNPとDonna Krasnewich, MD, PhDに感謝したい。 また、遺伝カウンセリングの専門家であるJenny Thies, MS, LGCに感謝する。
Funding: IJ ChangはNational Institutes of Health T32GM007454の支援を受けています。
脚注
利益相反(Conflicts of Interest)。 著者らは申告すべき利益相反はない。
Informed Consent:
- Jaeken J, Matthijs G. Congenital disorders of glycosylation(ヤーケン、マティス、先天性糖鎖異常症)。 Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family(先天性糖鎖異常症:急速に拡大する疾患群). Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. 先天性グリコシレーション障害Ia型(CDG-Ia)の原因となるPMM2における変異。 Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides(先天性糖鎖異常症:ドリコール結合オリゴ糖の生合成に影響を及ぼす欠陥に関する最新情報)。 Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. Population Carrier Frequencies and Diagnosed Patients based on Estonia in PMM2-CDG Prevalence of PMM2-CDG in Estonia. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. CDG-Ia (congenital disorders of glycosylation type Ia)で最も多く見られるPMM2変異に対するハーディーワインバーグ均衡の欠如。 European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome.新しく認識された遺伝的症候群におけるシアル酸欠乏血清および脳脊髄液トランスフェリン。 Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG nomenclature: time for a change! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways.糖鎖障害の解決に向けて:基本的なアプローチから複雑な経路を明らかにする。 Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis 2017;40:569-86。
- Cylwik B, Naklicki M, Chrostek L, et al. Congenital disorders of glycosylation.先天性グリコシレーション障害. 第1部 タンパク質N-グリコシル化の欠陥。 Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. サイエンス2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) 患者96人のフランスにおける臨床、検査、分子所見と長期追跡データおよび文献のレビュー. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. イタリアにおけるPMM2-CDGの全国調査:L32R変異に伴う軽度神経変形の高い頻度. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. 南アフリカにおけるALG6-CDG: 5人の新規患者の遺伝子型-表現型の記述。 JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. 異なる糖転移酵素は、分泌、二量化、自己糖化のために異なる処理をされる。 Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation(先天性糖鎖形成障害). で。 モラ-モンテスH.エディター。 糖鎖。 生化学、特性解析、応用 先天性糖鎖障害。 Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis.先天性糖鎖形成障害:魚鱗癬の他の原因。 Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. CDGにおける皮膚症状. J Inherit Metab Dis 2014;37:699-708。
- Miller BS, Khosravi MJ, Patterson MC, et al. 先天性糖鎖障害のある小児のIGF系. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presents in congenital disorders of glycosylation I: a series of 26 cases.先天性糖鎖異常症における幅広い臨床像。 J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Preubertal growth in congenital disorder of glycosylation type Ia (CDG-Ia)(先天性糖鎖異常症における思春期の成長)。 アーチディズチャイルド2002;87:324-7。
- Grünewald S, Imbach T, Huijben K, et al. 先天性糖化障害Ic型の臨床的および生化学的特徴、N型糖鎖合成における最初に認められた小胞体異常。 Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. 先天性糖鎖障害:その分子基盤、臨床像および特異的治療法のレビュー。 Eur J Pediatr 2003;162:359-79。
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–a recently discovered hereditary metabolic disease. 多臓器症状、発症率1/80,000、治療が困難である。 Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. 先天性糖鎖異常症Ia型の患者における肥大型心筋症による重症一過性心筋虚血症。 Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia)。 J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. 先天性糖鎖異常症Ia型における心嚢液および腹腔液貯留。 Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. 先天性糖鎖異常症Ia型(CDG-Ia)の成人における臨床的特徴。 Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. 炭水化物欠乏性糖蛋白質症候群。 Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. 1型シアロトランスフェリンパターンと多様な民族的起源を持つ患者における先天性糖鎖異常の臨床的および分子的特徴. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. ポティウイルスPlum pox virusのコートタンパク質のO-GlcNAc修飾はウイルス感染を促進する。 Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Scandinavian CDG-Ia patients: genotype/phenotype correlation and geographic origin of founder mutations.北欧のCDG-Ia患者、遺伝子型と表現型の相関と創始者変異の地理的起源。 Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 French adult patients with PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of the late-onset phenotype. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. glycosylation PMM2-CDGの先天性障害の網膜特性. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype.先天性グリコシル化障害(CDG-Ia)。 アーチディスチャイルド2001;85:236-9。
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. PMM2-CDGにおける甲状腺機能:診断アプローチと提案されたマネージメント。 Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J.成人の早期発症の小脳失調症を呈するIa型先天性糖質異常症。 Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype)(先天性糖質制限症候群Ⅰa患者における境界型の精神発達)。 J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype(1a型先天性糖鎖異常症:軽度の神経症状を有する3人の兄弟姉妹). J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. 糖質代謝の新しい障害:先天性糖鎖障害とその内分泌系への影響。 Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency.先天性糖代謝異常症は3人の幼児にみられる多臓器不全と低血糖の異質性。 J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. 小児科研究1995;37:395-401。
- Kristiansson B, Stibler H, Wide L. Gonadal function and glycoprotein hormones in the carbohydrate-deficient glycoprotein (CDG) syndrome(糖鎖欠乏症における生殖腺機能と糖鎖ホルモン). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib.(ホスホマンノースイソメラーゼ欠損症の臨床スペクトラム、CDG-Ibに対するマンノース治療の評価)。 Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. 先天性糖鎖異常症(CDG)の心臓合併症:文献の系統的レビュー. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. 炭水化物欠乏性糖タンパク質症候群に伴う再発性非免疫性胎児水腫症. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation(ホスホマンノースイソメラーゼ欠損:糖タンパク質欠損症候群、肝腸症状). Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib(糖鎖欠損性糖タンパク質症候群Ib)。 ホスホマンノースイソメラーゼ欠損症とマンノース療法。 J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. 糖鎖欠乏症状としての重症低血糖症。 J Pediatr 1999;135:775-81。
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Phosphomannose isomerase 欠損症における提示症状としての高インスリン血症低血糖症。 マンノースで治療可能な炭水化物欠乏性糖タンパク質症候群の新しい症状。 J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical Utility gene card for: ALG6 欠損型先天性糖鎖障害. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. 気道粘液: 生産から分泌まで。 Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. 新規GALNT3変異による高リン血性家族性腫瘍性石灰化症および高リン血性骨膜症症候群における長期臨床経過と表現型変動;症例報告および文献のレビュー. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparan sulfate in skeletal development, growth, and pathology: The case of hereditary multiple exostoses.骨格形成、成長、病理におけるヘパラン硫酸の役割について. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome.創始者効果によるホモ接合体p.R270C変異が、レユニオン島のラーセン症候群を引き起こした。 Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. 雑多な非炎症性筋骨格系疾患。 高リン酸血症性家族性腫瘍性カルシノーシス(FGF23、GALNT3、αKlotho)。 Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Galnt3遺伝子のアブレーションは、Fgf23の発現が増加しているにもかかわらず、低血中Fgf23濃度および高リン酸血症を引き起こす。 Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. ガングリオシド生合成酵素ST3GAL5の変異により、糖脂質と糖タンパク質のグリコシル化が変化した神経皮膚障害である塩&コショウ症候群が発症することを明らかにした。 Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. B4GALNT1 (GM2 synthase) の変異はガングリオシド生合成の新しい障害の根幹をなす. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA変異は早期発症のてんかん性脳症と特徴的な症状を引き起こす. Neurology. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN.「多発性先天異常-ハイポトニア-発作症候群はPIGNの変異によって引き起こされる。 J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. PIGMのhypomorphic promoter mutation causes inherited glycosylphosphatidylinositol deficiency.(PIGMのhypomorphic promoter mutationは遺伝性グリコシルホスファチジルイノシトール欠損症を引き起こす). Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. GPIアンカーリモデリング蛋白をコードするPGAP2のhypomorphic mutationsは常染色体劣性知的障害の原因となる。 Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. PIGAの生殖細胞変異の表現型:発作性夜間血色素尿症で体細胞変異する遺伝子. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. GPIアンカー合成経路のメンバーであるPIGOの変異は精神遅滞を伴う高ホスファターゼ症を引き起こす。 Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. PIGTのホモ接合体変異によるGPIアンカー欠損による新規知的障害症候群. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. グリコシルホスファチジルイノシトール遺伝子PIGLの変異はCHIME症候群を引き起こす。 Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. PIGA欠損症の遺伝子型と表現型スペクトラム。 Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene.PIG-A遺伝子の体細胞変異によるものである。 EMBO J 1994;13:110-7.
- Brodsky RA. 発作性夜間ヘモグロビン尿症(Paroxysmal nocturnal hemoglobinuria)。 Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation.先天性糖鎖異常症の診断における質量分析の影響. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. DOLK変異による常染色体劣性拡張型心筋症は、ジストログリカンのO-マンノシル化異常によるものであった。 PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distributions of exons and introns in the human genome.ヒトゲノムにおけるエクソンとイントロンの分布.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders.In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al.は、ヒトの全ゲノム配列決定による疾患の診断について述べた。 N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. ターゲット遺伝子シーケンスパネルと比較した診断収率の向上は、第一層の遺伝子検査として全ゲノムシーケンスの役割を示唆する。 Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. 遺伝性疾患が疑われる子どもに対するエクソームシーケンスのコストと診断収量:ベンチマーク研究。 Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics.「臨床エクソーム・ゲノムシーケンスの二次所見報告の推奨事項」, 2016 update. Genet Med 2017;19:249-55.
- Rothstein MA. 現代倫理の潮流。 GINA、ADA、そして雇用における遺伝的差別。 J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. 先天性糖鎖異常症(CDG)Ib型とマンノースによる治療を受けた小児の血栓塞栓症再発の一例. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. CDG-Ibの患者2名におけるマンノース治療にもかかわらずの肝疾患の発症。 Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice.マンノースサプリメントが胚性致死と失明を誘発する。 FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. CDG症候群1b型(MPI-CDG)患者におけるマンノース静脈内投与中の発作および昏迷。 J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. 白血球接着不全症II型におけるフコースサプリメント。 血液2000;95:3641-3。
- Almeida A, Layton M, Karadimitris A. Inherited glycosylphosphatidyl inositol deficiency: a treatable CDG.遺伝性グリコシルホスファチジルイノシトール欠損症。 Biochim Biophys Acta 2009;1792:874-80。
- Wong SY, Gadomski T, van Scherpenzeel M, et al. PGM1-CDGにおけるD-ガラクトース経口補給。 Genet Med 2017;19:1226-35。
- Morelle W, Potelle S, Witters P, et al. TMEM165-CDG患者におけるガラクトース補給は糖鎖欠損を救済する. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8欠損症:マンガン療法による生化学的補正と大きな臨床的改善。 Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. GNEミオパシーのマウスモデルにおける腎臓および筋肉の低シアリル化を逆転させる単糖類経口治療法. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al.CDG治療法。 From Bench to Bedside(ベンチからベッドサイドへ)。 Int J Mol Sci 2018;19:1304.
- Krasnewich D. Human glycosylation disorders. マリノPA、編集。 Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency(遺伝性GPI欠損症に対する標的治療). N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency.(ケトジェニックダイエット-ピガ欠損症による早期てんかん脳症の新規治療法). Brain Dev 2016;38:848-51.
.