- Overview
- Epidemiológia
- Biokémiai osztályozás és nomenklatúra
- Genetika
- Patofiziológia
- N-hez kötött fehérje glikozilációs hibák
- O-kötésű glikozilációs hibák és kombinált N- és O-kötésű glikozilációs hibák
- Lipidglikoziláció és GPI-horgony bioszintézisének hibái
- Klinikai manifesztációk
- N-hez kötött fehérjék glikozilációs hibái
- PMM2-CDG (CDG-Ia, PMM2-hiány)
- MPI-CDG (CDG-Ib, mannoszefoszfát izomeráz hiány)
- ALG6-CDG (glükozil-transzferáz 1 hiánya)
- O-kötött glikozilációs hibák és kombinált N- és O-kötött glikozilációs hibák
- Lipidglikoziláció és GPI-horgony bioszintézisének hibái
- Diagnózis
- N-hez kötött fehérje glikozilációs hibák
- O-kötésű glikozilációs hibák és kombinált N- és O-kötésű glikozilációs hibák
- Lipidglikozilációs és GPI-horgony bioszintézis hibák
- Molekuláris elemzés
- Kezelés
- Célzott terápiák és prognózis
- Köszönet
- Footnote
Overview
A glikoziláció a fehérjékhez és lipidekhez cukormaradványok hozzáadása a különböző sejtpályákon. A veleszületett glikozilációs rendellenességek (CDG) egy genetikailag és klinikailag heterogén, több mint száz betegségből álló csoport, amelyet a glikánszintézis vagy módosítási útvonalak különböző lépéseinek hibái okoznak. E monogén betegségek többsége autoszomális recesszív öröklődésű, de leírtak autoszomális domináns és X-kapcsolt formákat is.
A CDG-k jellemzően több rendszerben jelentkező manifesztációkkal, leggyakrabban fejlődési késéssel, gyengélkedési zavarral, hipotóniával, neurológiai rendellenességekkel, hepatopátiával és koagulopátiával járnak. Az érintett egyéneknél szem-, bőr- és szívbetegség, valamint arcdiszmorfizmus is előfordulhat. Bár az érintett egyének többségénél neurológiai elváltozások és kognitív késések figyelhetők meg, vannak olyan esetek, sőt típusok is, amelyeknél nem jelentkeznek neurológiai tünetek. Tekintettel a CDG széles klinikai és genetikai etiológiájára, a klinikai diagnózis a magas gyanúindexre támaszkodik a multiszisztémás betegségben.
A szérum szénhidráthiányos transzferrin (CDT) analízis az első vonalbeli szűrővizsgálat a CDG gyanúval rendelkező betegeknél, de kimutatása a szialinsavhiánnyal járó N-glikozilációs defektusokra korlátozódik. A következő vonalbeli vizsgálatok közé tartozik a dolicholhoz kötött glikánok elemzése és a genetikai vizsgálat. Az exponenciálisan növekvő betegségek e csoportjának korai diagnózisa fontos, mivel egyes CDG-k kezelhetőek. A glikozilációs hibák kezelése főként támogató jellegű, bár célzott terápiák állnak rendelkezésre az MPI-CDG, SLC35C1-CDG, PIGM-CDG és PGM1-CDG esetében. E kezelésekkel kapcsolatos részletek az alábbi “Célzott terápiák és prognózis” szakaszban találhatók. Ez az áttekintés a CDG leggyakoribb, felismerhető fenotípussal vagy kezeléssel rendelkező típusaira összpontosít, a célközönség pedig az alapellátók.
Epidemiológia
A CDG összes típusának előfordulása és prevalenciája összességében nem jól meghatározott, bár világszerte szinte minden etnikai háttérből jelentettek betegeket, és mindkét nemet egyformán érinti. Az európai és afroamerikai populációkban a becsült prevalencia 1/10 000-re tehető az 53 gén ismert patogén variánsainak hordozói gyakorisága alapján (1-4). A leggyakrabban diagnosztizált CDG, a PMM2-CDG prevalenciája a holland populációkban 1/20 000, Észtországban pedig 1/77 000 között mozog az elszigetelt jelentések alapján (5,6). A mai napig a legtöbb CDG-típus esetében 100-nál kevesebb esetet jelentettek.
Biokémiai osztályozás és nomenklatúra
Tágabb értelemben a CDG-ket jelenleg négy kategóriába sorolják – (I) N-hez kötött glikoziláció, (II) O-hez kötött glikoziláció, (III) kombinált N- és O-hez kötött/többszörös glikoziláció, és (IV) lipid- és glikozil-foszfatidil-inozitol (GPI) horgony bioszintézis hibák.
A PMM2-CDG, korábban Ia típusú CDG néven ismert N-hez kötött fehérje glikozilációs hiba volt az első CDG, amelyről Jaeken számolt be 1980-ban, és a mai napig messze a leggyakoribb CDG (7). A PMM2-CDG-t kezdetben “szénhidráthiányos glikoprotein szindrómának” nevezték el, mivel az érintett egyéneknél a szérum transzferrin izoelektromos fókuszálásával többszörös szérum glikoprotein rendellenességeket észleltek. Korábban a CDG-t a transzferrin izoforma analízisének mintái alapján osztályozták – az I-es típusú mintákat a citoplazmában vagy az ER-ben lokalizálódó, doliholhoz kötött glikánok összeszerelési és transzferhibáinak tulajdonították, a II-es típusú mintákat pedig a Golgi-apparátus feldolgozási hibáinak tulajdonították. Ettől az elágazási ponttól kezdve a CDG-ket a felfedezés sorrendjében, ábécésorrendben nevezték el.
A széles körű molekuláris diagnosztika megjelenésével a CDG-nómenklatúrát 2008-ban frissítették a betegség molekuláris etiológiájának meghatározása érdekében, tükrözve a korábban kialakított dichotóm kategóriákba nem illeszkedő kórokozók és rendellenességek exponenciális növekedését. Jelenleg a CDG-nómenklatúrát az érintett gén neve jelöli (nem ékezetes, génnevek a www.genenames.org oldalon), amelyet -CDG követ (pl. PMM2-CDG) (8).
Genetika
A veleszületett glikozilációs rendellenességek túlnyomó többsége autoszomális recesszív módon öröklődik, mindkét tünetmentes (hordozó) szülőtől egy-egy mutáció öröklődik. A genetikai diagnózis felállításához molekuláris vizsgálatra van szükség, általában újgenerációs szekvenálási módszerekkel. Az ismert variánsra vonatkozó szülői vizsgálat megerősítheti az öröklődést a de novo előfordulással szemben. Autoszomális recesszív öröklődés esetén a testvérek és az érintett egyén minden egyes terhessége esetén a kiújulás kockázata 25%, ha érintett, 50%, ha tünetmentes hordozó, és 25%, ha nem érintett.
Egy maroknyi CDG autoszomális domináns öröklődésű (N-hez kötött: GANAB-CDG, PRKCSH-CDG; O-kapcsolt: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Kevesebb X-kapcsolt (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). A CDG legtöbb domináns és néhány X-kapcsolt formája de novo mutációkra vezethető vissza. A konkrét betegségeket és géneket az alábbiakban a “patofiziológia” részben ismertetjük.
A CDG összes publikált génjének mutációs adatai elérhetők a Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) adatbázisban. A specifikus génváltozatokra vonatkozó információk a Leiden Open Variation Database integrált in silico patogenitási eszközökkel ellátott adatbázisában (http://www.lovd.nl/3.0/home) érhetők el. Az egyes génekre vonatkozó klinikai összefoglalók megtalálhatók az Online Mendelian Inheritance in Man (http://www.omim.org/) vagy szűkebb körben a GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/) oldalon. Tekintettel az érintett betegek kis számára a legtöbb CDG altípus esetében, a genotípus-fenotípus korrelációt nehéz megállapítani.
Patofiziológia
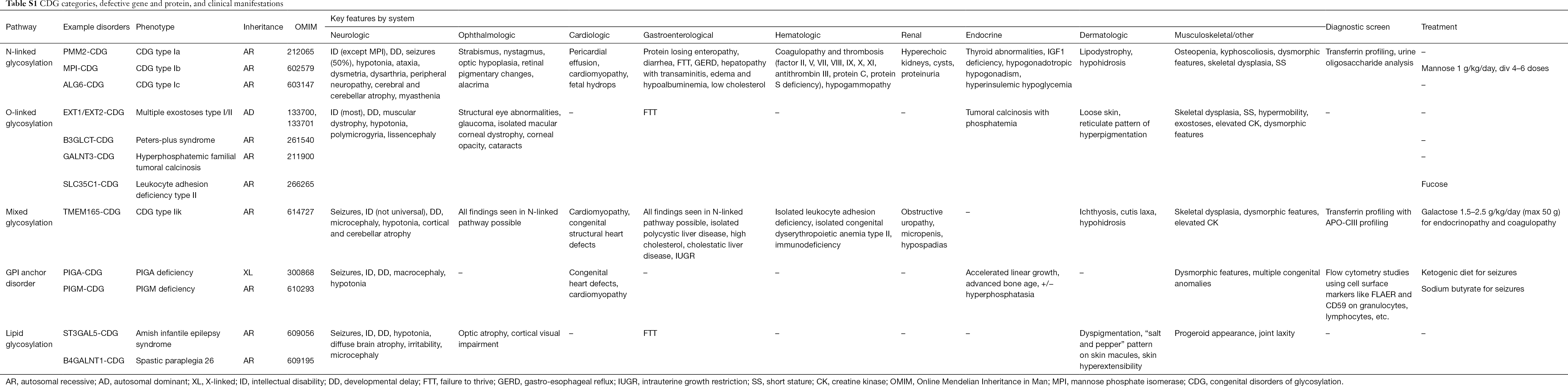
A CDG több mint 130 típusáról számoltak be eddig (9,10). Tekintettel a glikozilációs útvonalak ubiquitárius jelenlétére, a CDG-k biokémiai patogenezise rendkívül változatos. Számos fehérje és lipid (pl. szfingolipidek és glikolipidek) glikozilálódik monoszacharidokkal és/vagy oligoszacharidokkal, amelyeket együttesen glikánoknak neveznek, a különböző sejtkompartmentekben. Szubcelluláris elhelyezkedésük változatos, de a legtöbb hiba az ER-ben vagy a Golgi-apparátusban fordul elő. A gyakoribb CDG klinikai jellemzőit és genetikai etiológiáját útvonalak szerint az S1. táblázat foglalja össze.
A fehérjék között a glikánokat a polipeptidlánchoz való kapcsolódásuk alapján írják le: az N-glikánok az aszparagin (Asn) amidcsoportjához, míg az O-glikánok a szerin vagy treonin hidroxilcsoportjához kapcsolódnak. Az N-glikán szintéziséhez nukleotidhoz kötött cukrok fokozatos felépítésére van szükség a citoszolban, az endoplazmatikus retikulumban történő összeszerelésre és a Golgi-készülékben történő feldolgozásra. Ezzel szemben az O-glikán szintézis összeszerelést igényel, de feldolgozást nem, ezért az O-glikozilációs hibák túlnyomórészt a Golgi-apparátusban fordulnak elő.
N-hez kötött fehérje glikozilációs hibák
Az N-glikoziláció magában foglalja a szénhidrátstruktúrák kovalens kötését az Asn-maradékok oldallánc-amidcsoportjához egy konszenzusos Asn-X-Ser/Thr akceptorhelyen belül, a szubsztrát polipeptid transzlokációját az endoplazmatikus retikulumba átalakítás céljából, és az N-glikán lánc további módosítását a Golgiban (11,12). A szintézis, az összeszerelés és a feldolgozás útvonalán bárhol előforduló hibák klinikai betegséghez vezethetnek.
A PMM2-CDG-t a foszfomannomutáz 2 (PMM2) gén patogén variánsai okozzák, ami a PMM2 enzim hiányához vezet, amely a guanozin-difoszfát (GDP) mannóz-szintézis második lépésében a mannóz-6-foszfát citoszolikus átalakítását mannóz-1-foszfáttá katalizálja. A legtöbb beteg összetett heterozigóta patogén miszense mutációt hordoz (www.lovd.nl/PMM2). A leggyakoribb visszatérő patogén p.Arg141His variáns az európai felmenőkkel rendelkező érintett egyének körülbelül 40%-ánál fordul elő, és a p.Phe119Leu is gyakran megtalálható Észak-Európában (1). A PMM2-CDG esetében genotípus-fenotípus korrelációkról számoltak be (3,13,14).
Az MPI-CDG egy autoszomális recesszív rendellenesség, amelyet a mannóz-foszfát izomeráz (MPI) gén patogén variánsai okoznak, ami hiányos foszfomannóz izomerázhoz (MPI) vezet. Az MPI normális esetben a GDP-mannóz szintézis első lépését katalizálja (azaz a fruktóz-6-foszfát átalakítását mannóz-6-foszfáttá), de a fruktóz-6-foszfát nem halmozódik fel intracellulárisan, mivel a glikolitikus útvonalon is metabolizálható. Ezért, bár biokémiailag hasonló a PMM2-CDG-hez, az MPI-CDG nem okoz olyan jelentős neurológiai és multiszisztémás érintettséget. A CDT a választott szűrővizsgálat az MPI-CDG esetében is, amely 1-es típusú mintázatot mutat. A diagnózist ezután molekulárisan vagy fibroblaszt/leukocita MPI-aktivitással lehet megerősíteni.
Az ALG6-CDG egy recesszív betegség, amelyet az ALG6 mutációi okoznak, ami három glükózmolekula kóros kötődését eredményezi a doliholhoz kötött mannóz intermedierekhez és a szérum glikoproteinek downstream hipoglikozilációját (15).
O-kötésű glikozilációs hibák és kombinált N- és O-kötésű glikozilációs hibák
Az O-glikoziláció a fehérjék szerin-, treonin- és hidroxilizin-maradványaihoz történő szénhidrátláncok fokozatos hozzáadását foglalja magában a Golgi-apparátusban található glikoziltranszferázok által (16). Az O-kötött glikánok számos típusát hozták összefüggésbe emberi betegségekkel, amelyeket az aminosavmaradványhoz kapcsolódó első cukor alapján neveztek el (17).
Lipidglikoziláció és GPI-horgony bioszintézisének hibái
A GPI-horgonyok olyan glikolipidek, amelyek az endoplazmatikus retikulumban szekvenciális összeállításon és a Golgiban történő módosításokon mennek keresztül. Az enzimhiányból eredő GPI-horgony bioszintézis rendellenességeket a felfedezés sorrendjében, nem pedig időrendi sorrendben, az összeszerelés lépései szerint nevezzük meg. A szintézis után a GPI-horgonyok a plazmamembránokon tartózkodnak, és több száz sejtfelszíni fehérjéhez kötődnek, számos sejtfunkciót ellátva. E betegségek többsége autoszomális recesszív, figyelemre méltó kivétel az X-kapcsolt PIGA-hiány.
Klinikai manifesztációk
A glikozilációs útvonalak ubiquitását tekintve gyakorlatilag bármely szervrendszer érintett lehet a CDG-ben, bár a legtöbb esetben neurológiai rendellenességekről van szó. Néhány CDG ichthyosisban jelentkezik, beleértve az MPDU1-CDG-t, a DOLK-CDG-t, az SRD5A3-CDG-t (1. ábra) és a PIGL-CDG-t (18,19). Majdnem minden CDG multi-szisztémás betegséggel jelentkezik az élet első néhány évében, kivéve néhányat, amelyek csak egyetlen szervrendszert érintenek (pl, retina a DHDDS-CDG-ben; neuromuszkuláris csomópont az ALG2-CDG-ben, ALG14-CDG-ben, CFPT1-CDG-ben; agy az ST3GAL3-CDG-ben, TUSC3-CDG-ben; bőr vagy vázizom a POGLUT1-CDG-ben, POFUT1-CDG-ben; porc az EXT1/EXT2-CDG-ben; máj a TMEM199-CDG-ben; vörösvértestek a SEC23B-CDG-ben). A betegség kialakulásának kora és súlyossága az újszülöttkori halálos kimenetelűtől a majdnem tünetmentes felnőttkorig, és a kettő közötti bármely permutációig terjedhet. A leggyakrabban jelentett tünetegyüttes a fejlődési késés, a gyarapodás elmaradása, hipotónia, neurológiai rendellenességek, hipoglikémia, valamint változó máj-, szem-, bőr-, gasztrointesztinális, immunológiai, csontváz- és véralvadási rendellenességek (19).
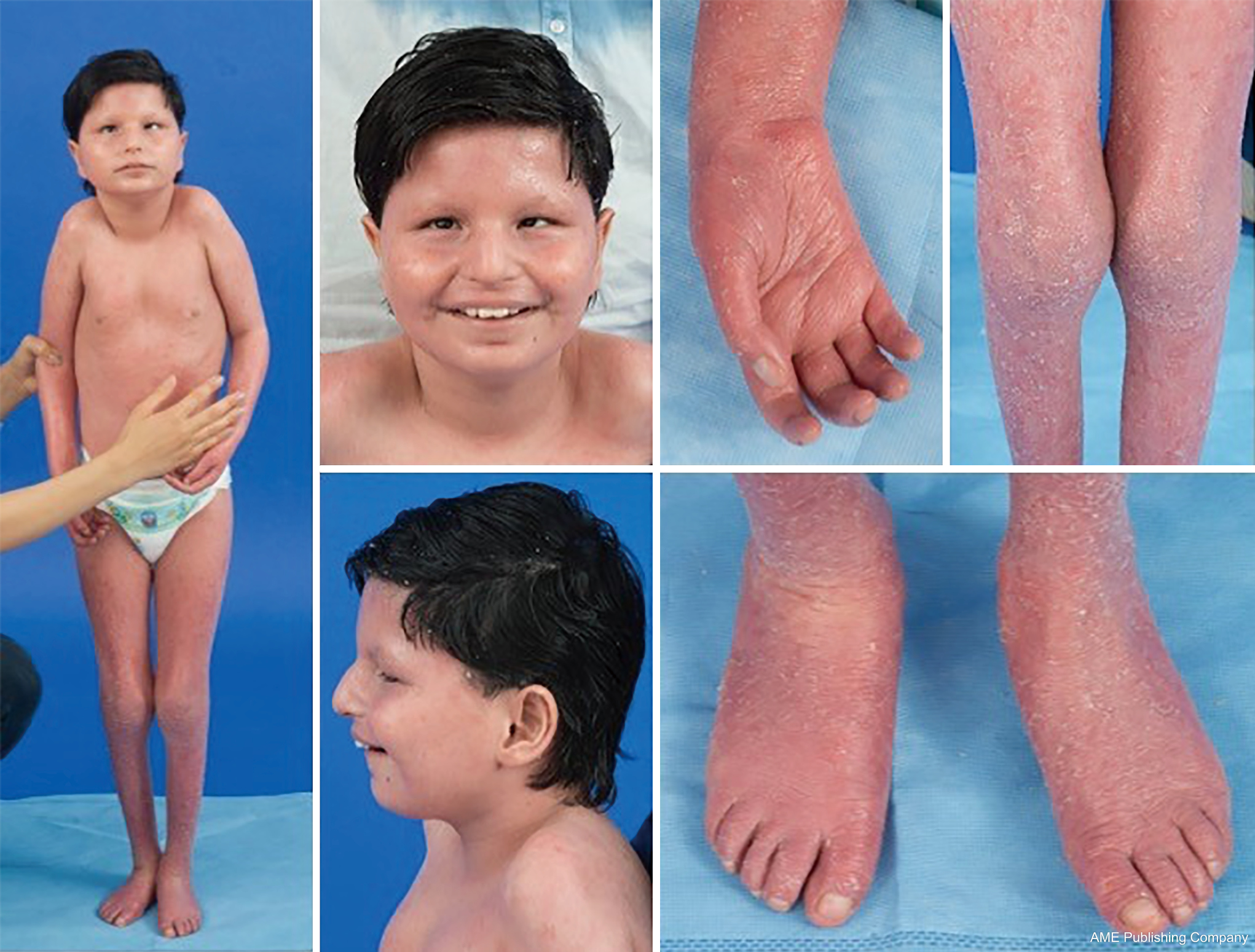
A bejelentett esetek ritkasága miatt számos CDG altípus teljes fenotípusa még nem teljesen körülhatárolt. Ezért a CDG-t a multiszisztémás betegségek bármelyikében figyelembe kell venni, különösen a neurológiai komponenssel vagy nem specifikus, tisztázatlan etiológiájú fejlődési késéssel járó esetekben.
Noha a multiszisztémás tünetek patofiziológiája még tisztázásra vár, tisztázódott a kapcsolat bizonyos glikozilációs útvonalak és a specifikus klinikai tünetek között. Például a CDG számos típusában megfigyelhető sikertelenség az inzulin növekedési útvonalán belül számos glikoprotein, köztük az IGF-1, ALS és IGFBP-3 hipoglikozilációjának és károsodott képződésének tulajdonítható (20). Ahogy egyre jobban megértjük a komplex rendellenességek e csoportját, a CDG-t egyre gyakrabban ismerik fel a nehezen felismerhető diagnózisú egyénekben. A különböző CDG-k szervrendszeri érintettségét az S1. táblázat foglalja össze. A CDG leggyakoribb és célzottan kezelt formáinak klinikai jellemzőit az alábbiakban tárgyaljuk.
Teljes táblázat
N-hez kötött fehérjék glikozilációs hibái
A leggyakrabban diagnosztizált CDG-ként az N-hez kötött glikozilációs zavarok fenotípusát gyakran klasszikus megjelenési formaként hirdetik. A CDG fenotípusos spektruma azonban igen változatos, és sok CDG nem a PMM2-CDG-hez társuló sztereotip tünetekkel jelentkezik.
PMM2-CDG (CDG-Ia, PMM2-hiány)
A PMM2-CDG a leggyakoribb CDG, világszerte több mint 700 esetet jelentettek. Csecsemőkorban multiszisztémás súlyos betegség, gyermekkorban neurológiai betegség és fejlődési késés és/vagy felnőttkorban stabil értelmi fogyatékosság jellemzi (21,22).
A PMM2-CDG csecsemőkorban jellemzően röviddel a születés után neurológiai rendellenességekkel jelentkezik, nevezetesen strabismus és rendellenes szemmozgások, kisagyi hypoplasia, hypotonia, pszichomotoros retardáció, ataxia, hypotonia és hyporeflexia. A csecsemőknél májbetegség, nefrotikus szindróma és veseciszták, perikardiális folyadékgyülem és hipertrófiás kardiomiopátia, sikertelenség és többszervi elégtelenség is előfordulhat, ami az érintettek akár 20%-ánál az első életévben halálhoz vezet (21,23-28).
A PMM2-CDG-ben szenvedő betegeknél a diszmorfikus jellemzők konstellációját írták le (2,3. ábra). Ezek közé tartozik a hipoplasztikus kisagy, az arc diszmorfizmusa (pl. nagy, diszplasztikus fülek), a kifordult mellbimbók és a zsírszövet abnormális eloszlása a fenék vagy a szuprapubikus régió felett, ami az életkor előrehaladtával megszűnhet (14,21,29-32). A betegek a leírások szerint kifelé forduló és vidám viselkedésűek. A betegség megjelenése igen változatos, bár a kancsalság az érintett betegek több mint 70%-ánál megfigyelhető (21,23,33-35). A betegek mintegy 25-50%-ánál fordított mellbimbó és rendellenes zsírpárna látható (36).

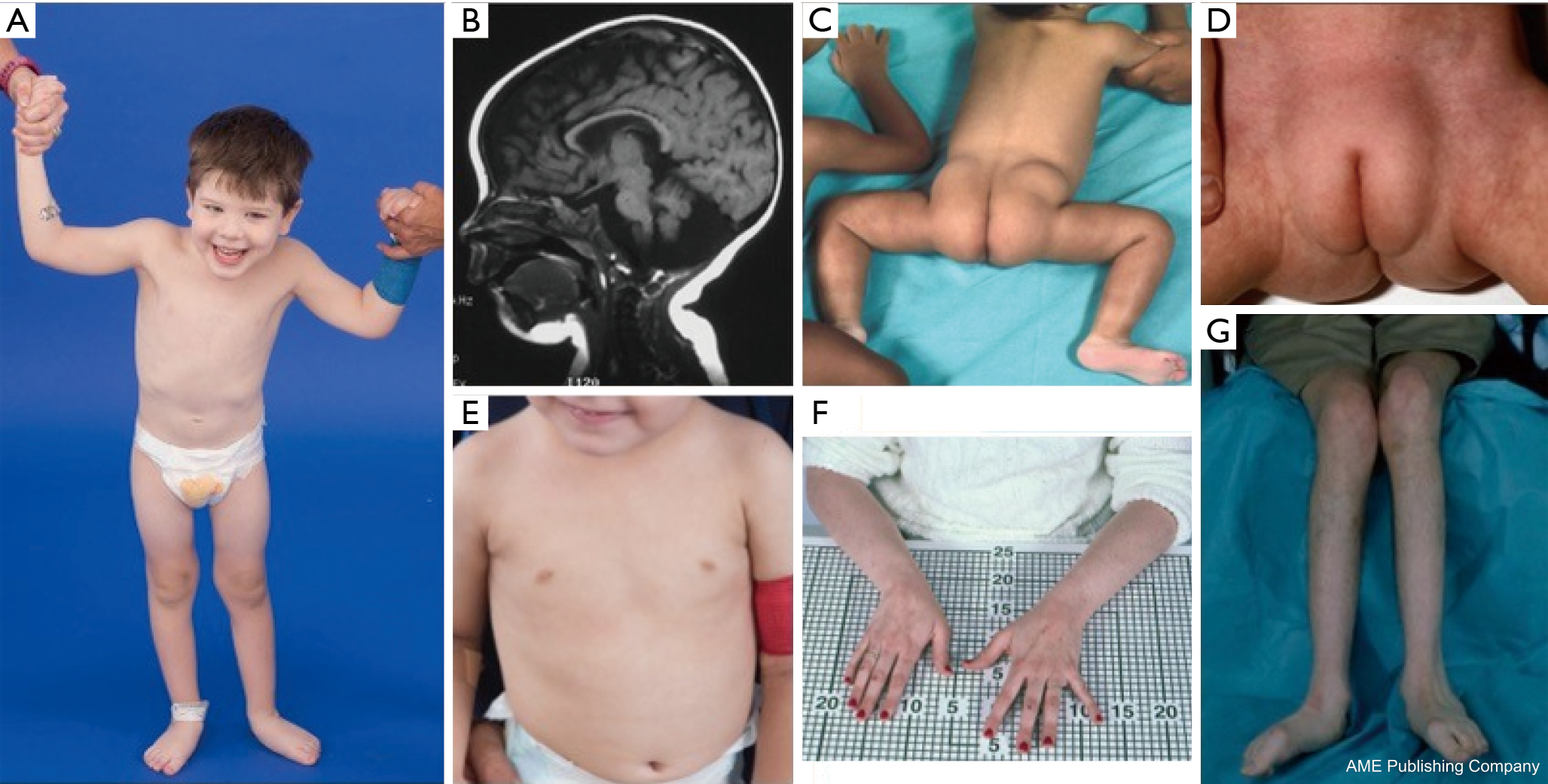
Gyermekkorban az érintett egyéneknél retinitis pigmentosa, stroke-szerű epizódok és rohamok, beszéd- és mozgáskésés, valamint perifériás neuropátia alakulhat ki. Konstitúciósan a betegeknél a táplálkozási és GI-rendellenességek és a globális fejlődési elmaradás miatt gyakran előfordul a gyarapodás elmaradása. Megemelkedett májtranszaminázok figyelhetők meg klinikai következmény nélkül, amelyek jellemzően 5 éves korig normalizálódnak, betegséggel járó alkalmi ingadozásokkal (21,24). Májbiopszia ritkán javallott CDG-ben, kivéve, ha májfibrózis gyanúja merül fel (1). A klinikai hypothyreosis ritka, de a CDG-ben szenvedő betegeknél meg kell mérni a pajzsmirigyhormonokat és a szabad T4-et, ami alacsony pajzsmirigy-kötő globulint (TBG) és a pajzsmirigy-stimuláló hormon (TSH) átmeneti emelkedését mutathatja (37). Máj- és epeúti malformációkról nem számoltak be PMM2-CDG-s betegeknél.
A PMM2-CDG-ben szenvedő felnőttek a 7. vagy 8. évtizedig élhetnek stabil kognitív késéssel, perifériás neuropátiával és progresszív mellkasi és gerincvelői kyphoscoliosisszal, osteopeniával vagy osteoporosisszal (34). A kisagyi ataxia egyre inkább felismert tünet a multiszisztémás érintettség mellett (38-40). Endokrin rendellenességek, beleértve a hyperprolactinaemiát, a növekedési hormon felszabadulását hyperglykaemiával, inzulinrezisztenciával és hyperinsulinémiás hipoglikémiával (41,42). Az érintett nőknél a hipogonadotrop hipogonadizmus a másodlagos nemi fejlődés hiányához vagy a petefészek hiányához vezethet (41,43,44). A betegeknél a csökkent szérum véralvadási faktorok, köztük a IV, IX és XI faktor, az antitrombin III, a C-protein és az S-protein csökkenése miatt fokozott lehet a trombózis kockázata (29).
MPI-CDG (CDG-Ib, mannoszefoszfát izomeráz hiány)
AzMPI-CDG egyedülálló, mert az érintett betegeknél kevés vagy egyáltalán nincs neurológiai érintettség, és a betegség egyes megnyilvánulásai orális mannózzal kezelhetőek (2). A tünetek főként hepatikus-bélrendszeri jellegűek, diszmorfikus jellegzetességek vagy kognitív késések nélkül. A betegek jellemzően visszatérő hányással, jelentős hipoglikémiával, gyengélkedési zavarral, potenciálisan életveszélyes fehérjepazarló enteropátiával, májfibrotikus elváltozásokkal és epevezeték-tágulattal jelentkeznek (45-51). A betegek a C- és S-protein, valamint az antitrombin III alacsony szérumkoncentrációja miatt a trombotikus események fokozott kockázatának vannak kitéve.
ALG6-CDG (glükozil-transzferáz 1 hiánya)
AzALG6-CDG a második leggyakoribb N-glikozilációs hiba, amelyet a PMM2-CDG-hez hasonló, de enyhébb fenotípus jellemez. Az ALG6-CDG-ben szenvedő betegeknél a gyarapodás elmaradása, fejlődési késés, hipotónia, görcsök, kancsalság, ataxia, koagulopátia és arcdiszmorfizmus (pl. alacsonyan ülő fülek, hipertelorizmus és makroglossia) fordul elő. Az MPI-CDG-hez hasonlóan fehérjevesztéses enteropátia is előfordulhat. Ezenkívül az érintett betegeknél csontrendszeri rendellenességek is előfordulhatnak, beleértve a brachydactylia és az ujjak malformációit, valamint a skoliózist. Az érintett betegeknek jellemzően nincs retinitis pigmentosa vagy kisagyi hipoplázia (52).
O-kötött glikozilációs hibák és kombinált N- és O-kötött glikozilációs hibák
Az O-glikánok jelentős jelenléte miatt a mucintartalmú fehérjékben, beleértve a glikozaminoglikánokat (GAG-okat) és a hámfelszíneket (53), a GAG-szintézis zavarai jellemzően csontvázdiszpláziákhoz vagy kötőszöveti betegségekhez vezetnek. Az érintett betegek neurológiai tünetek mellett mozgásszervi, bőr- és ízületi eltérésekkel (pl. ízületi laxitás, multiplex exostosisok, chondro/osteosarcomák) is jelentkezhetnek (54-56). Például az N-acetilgalaktozaminitranszferáz 3 (GALNT3) O-glikozilálja a foszfaturikus hormont, az FGF23-at, megakadályozva a proteolitikus hasadást és lehetővé téve az intakt szekréciót. A GALNT3-hiány familiáris tumoros kalcinózishoz vezet, amelyet hiperfoszfatémia és ektopikus meszesedés jellemez (57,58).
Lipidglikoziláció és GPI-horgony bioszintézisének hibái
A glikoszfingolipideket és szialilált származékaikat, a gangliozidokat elsősorban a neuronok expresszálják. A gangliozidok lebontásának hibái felhalmozódáshoz és a jól jellemzett lizoszomális tárolási betegségekhez vezetnek. Ezzel szemben a gangliozidok bioszintézisének hibái, mint például az ST3GAL5-CDG és a B4GALNT1-CDG, rendkívül ritkák, és súlyos neurodegeneratív betegségekhez vezetnek. A betegek spasztikus parapláziával, súlyos értelmi késéssel, epilepsziával és nem neurológiai tünetekkel, köztük csontvázdiszpláziával, diszmorfiával és rendellenes bőrpigmentációval jelentkezhetnek (59,60).
A GPI-horgony bioszintetikus útvonalán belül számos gén mutációja számos veleszületett rendellenességet, értelmi fogyatékosságot és epilepsziát okoz. A legjobban jellemzett GPI bioszintézis defektus, az X-hez kötött PIGA hiány, csecsemőkori görcsökkel, hipszarrhythmiával, hipotóniával, többféle agyi rendellenességgel és arcdiszmorfiával jár. A betegeknél változó bőr-, máj-, szív- és vesebetegség is előfordulhat (61-69). A PIGA egyes mutációi a fenotípusosan elkülönülő betegséget, a paroxysmalis nocturnalis hemoglobinuriát (PNH) okozzák, amely a csontvelő elégtelenségével járó szerzett rendellenesség (70,71).
Diagnózis
Ha a CDG klinikai gyanúja felmerül, az első lépés a plazma vagy szérum biokémiai CDG-vizsgálatának elrendelése, beleértve a CDT és az N-glikán vizsgálatát. A szérum CDT és N-glikán analízis csak az N-glikozilációs hibákat képes kimutatni, ezért nem lenne hasznos az izolált O-glikozilációs vagy GPI-horgonyzási hibák megkülönböztetésében. A transzferrin izoforma-elemzést eredetileg a transzferrin izoelektromos fókuszálásával végezték, mivel az N-glikán szintézis elmaradása a szialinsav részleges hiányát okozza, ami megváltoztatja a szérum transzferrin töltését, majd katódos vándorlását az elektroforetikus mezőn. A transzferrin és az N-glikán tömegspektrometrián alapuló elemzése azonban mára nagyrészt felváltotta az izoelektromos fókuszálást az oligoszacharidok tömeg és töltés szerinti specifikus változásainak azonosításával (72).
N-hez kötött fehérje glikozilációs hibák
A szérum transzferrin CDT eredményeit a mono-oligoszacharid/di-oligoszacharid transzferrin arányaként közlik, a-oligoszacharid/di-oligoszacharid transzferrin, tri-sialo/di-oligoszacharid transzferrin, apolipoprotein CIII-1/apolipoprotein CIII-2 és apolipoprotein CIII-0/apolipoprotein CIII-2 aránya. Ezek a kvantitatív eredmények a leletek mintázatának értelmezésével is együtt járnak.
Az I. típusú mintázatú transzferrin CDT-t fokozott di- és aszialotranszferrin sávok jellemzik, és a citoszolban vagy az endoplazmatikus retikulumban az N-glikánszintézis hibáira utal. A II-es típusú mintázatot megnövekedett di- és asialotranszferrin sávok, valamint tri- és/vagy monoszialotranszferrin sávok jellemzik, és az N-glikánok Golgi-apparátusban történő feldolgozásának hibáira utal (73).
Ha I-es típusú szérum transzferrin CDT mintázatot észlelünk, a PMM2- vagy MPI-hiánynak a differenciáldiagnózisok előterében kell állnia, mivel a PMM2-CDG a leggyakoribb CDG, az MPI-CDG pedig kezelhető és kezeletlenül potenciálisan végzetes lehet. A diagnózisok megkülönböztetése érdekében N-glikán profilalkotást, molekuláris szekvenálást vagy enzimatikus vizsgálatot kell végezni. A PMM2-CDG vagy MPI-CDG diagnózisát a PMM2 vagy MPI biallelikus patogén variánsait kimutató molekuláris vizsgálatokkal, majd a leukocitákban vagy fibroblasztokban kimutatott PMM vagy MPI enzimaktivitással kell megerősíteni, ha a genetikai variánsok patogenitása bizonytalan. Az N-glikán analízis vagy a molekuláris analízis az ALG-CDG többségét megkülönböztetné a PMM2 vagy MPI-CDG-től (15).
A II. típusú szérum transzferrin CDT mintázat Golgi defektusokra, például N-acetilglükozaminil-transzferáz (GnT) II hiányra utal (CDG IIA típus, MGAT2-CDG). Az Apolipoprotein CIII (Apo-CIII) izoforma-analízis a II-es típusú CDT-profil kiegészítő vizsgálata, mivel a Golgi-apparátusban a mucin típusú O-glikozilációs hibákat méri. A CDT vagy az Apo-CIII korlátozott érzékenységű a II. típusú CDG kimutatásában. Ezért N-glikán és O-glikán profilalkotást és molekuláris panel vagy exom szekvenálást kell végezni, ha ezek a klinikai tesztek rendelkezésre állnak. A transzferrin-glikozilációs mintázatok sporadikusan normalizálódhatnak; ezért a magas gyanúindexű betegeknél ismételt vizsgálat indikálható. Hamis pozitív eredményt kaphatunk az örökletes fruktóz-intolerancia akut krízisében, galaktosémiában, akut májbetegségben és egyes bakteriális fertőzésekben szenvedő betegeknél. A biokémiai CDG-tesztek egyike sem alkalmas az összes CDG szűrésére, ezért még normális szűrési eredmények esetén is molekuláris génpanel-vizsgálat vagy exom-szekvenálás végezhető erős klinikai gyanú esetén. Ezzel szemben a molekuláris genetikai leletek biokémiai és funkcionális megerősítése is elengedhetetlen, mivel a CDG-ben szenvedő betegek többsége legalább egy enyhe és gyakran újszerű miszenzmutációt hordoz.
O-kötésű glikozilációs hibák és kombinált N- és O-kötésű glikozilációs hibák
A diagnózis a molekuláris szekvenálásra támaszkodik, mivel a transzferrin izoforma-elemzés nem mutatná ki az izolált O-glikozilációs hibákat. A kombinált N- és O-kötött glikozilációs hibák CDT-vel, ApoCIII analízissel, valamint plazma N- és O-glikán analízissel kimutathatók.
Lipidglikozilációs és GPI-horgony bioszintézis hibák
A vér granulocitáinak áramlási citometriája a GPI-horgonyzott fehérjék, például a CD16 és CD24 sejtfelszíni expresszióját méri. A fehérvérsejtek vagy vörösvérsejtek áramlási citometriás elemzése bizonyos GPI-horgonyzott sejtfelszíni fehérjékre vonatkozóan klinikailag elérhető a PIGA gén szerzett mutációi miatt kialakuló PNH tesztjeként. A PNH-teszt más GPI-horgonyzási hiányosságok rendellenességeit is kimutatja, de a diagnózis többnyire molekuláris elemzésre támaszkodik.
Molekuláris elemzés
A CDG legnagyobb diagnosztikai hozamát az újgenerációs alapú génszekvenáló panel vagy klinikai exomszekvenálás (CES) adja. A génszekvenálás a gének nukleotidszekvenciáját, vagyis “betű szerinti” írásmódját ellenőrzi annak megállapítására, hogy van-e olyan változás, amely befolyásolja a gén működését. Az emberi genom 3 millió nukleotidból áll, de ennek csak 1-2%-át, az úgynevezett exonokat fordítják funkcionális fehérjetermékké. A fennmaradó, az exonok között elhelyezkedő, nem kódoló DNS-t, amelyet nem fordítanak le, intronoknak nevezzük (74). A CES az emberi genom mintegy 20 000 génjének szinte valamennyi ismert exonját vizsgálja, amelyek a kromoszómák genetikai anyagának kisebb részét teszik ki, de a legnagyobb valószínűséggel betegséget okozó (patogén) variánsokat tartalmaznak. A CES magában foglalhatja a mitokondriális DNS (mtDNS) szekvenálását is, amely a mitokondriumokban található kis, extranukleáris, körkörös DNS-t kérdezi le, amely kizárólag anyai öröklődésű.
A CES lehetséges eredményei között pozitív, negatív és ismeretlen jelentőségű változatok is szerepelnek. A pozitív eredmény azt jelenti, hogy ismert betegséget okozó (azaz patogén) variánsokat azonosítanak, ami után a diagnózis, a természetes lefolyás, a prognózis, a kiújulás kockázata és a kezelési lehetőségek megvitathatók. A negatív eredmény azt jelenti, hogy nem azonosítottak kimutatható patogén variánsokat. Az ismeretlen jelentőségű variánsok (más néven VUS) azt jelenti, hogy bár genetikai változásokat azonosítottak, nincs elég információ a konkrét genetikai változásról ahhoz, hogy véglegesen meg lehessen állapítani, hogy az betegséget okoz-e. A VUS azt jelenti, hogy az adott genetikai változás nem okoz-e betegséget. Az egyes személyek DNS-ében várhatóak eltérések, ezért a szülői minták egyidejű vizsgálata az összehasonlítás érdekében segítheti az eredmények laboratóriumi és klinikai értelmezését. A CES diagnosztikai hozamát 30-35%-ra becsülik, és az idő múlásával a génfelfedezés és az emberi genommal kapcsolatos ismeretek folyamatos fejlődésével egyre nő (75-77). A CES-t egyre gyakrabban rendelik első vonalbeli, széles körű genetikai vizsgálatként, mivel gyors átfutási idővel és az elemzett genetikai információ mennyiségéhez képest alacsony relatív költséggel jár. A CES korlátai közé tartozik a 100%-os érzékenység hiánya, a genetikai változások bizonyos típusainak (pl. deléciók, duplikációk, trinukleotid ismétlődések, mély intronikus mutációk vagy metilációs hibák) kimutatására való képtelenség, valamint az a tény, hogy a diagnózis nem feltétlenül nyújt további információt a betegségről vagy a kezelés megváltoztatásáról.
A CES jelentésében a jól ismert genetikai állapotokhoz kapcsolódó génekben véletlenül felfedezett patogén variánsok másodlagos leletként szerepelhetnek (78). Az ajánlott betegségek e listáját az American College of Medical Genetics (ACMG) kurátora állította össze. A genetikai információk diszkriminációmentességéről szóló törvény (GINA) fontos szempont annak eldöntésekor, hogy a véletlen leletek megismerését választják-e vagy sem (79). A GINA védi az egyéneket a genetikai információkkal való visszaéléstől az egészségbiztosítás és a foglalkoztatás terén, de az életbiztosítást nem. A GINA a következő genetikai információkat védi: családi kórtörténet, hordozóvizsgálat, prenatális genetikai vizsgálat, fogékonysági és prediktív vizsgálatok, valamint a tumorok elemzése vagy más, gén-, mutáció- vagy kromoszóma-változásokra vonatkozó értékelések.
Kezelés
A CDG kezelése nagyban függ az egyén specifikus tüneteitől. A CDG-ben szenvedő betegek visszatérő tünetei közé tartozik a gyarapodás elmaradása, a globális fejlődési késés, a hányás, a stroke-szerű epizódok és a csontrendszeri rendellenességek. A klinikai vagy szubklinikai koagulopátia, endokrinopátia, hepatopátia és szívhibák szintén gyakran előfordulnak. A betegség mértékének megállapítására alapszintű laboratóriumi vizsgálatok és rutinszerű megfigyelés javasolt, különösen a PMM2-CDG esetében. Ezek közé tartoznak a májfunkciós vizsgálatok, a szérum albumin, a pajzsmirigyfunkciós vizsgálatok, beleértve a szabad T4-et, a protein C, protein S, antitrombin III, IX faktor, vizeletvizsgálat, valamint a szérum gonadotropinok és a növekedési hormon.
A képalkotó vizsgálatok közül ajánlott az echokardiogram, a veseultrahang, a csontkorszak, a szemészeti vizsgálat a lencse, a retina, a szemmozgás és az intraokuláris nyomás értékelésére. Hacsak másként nem jelzik, a CDG-ben érintett felnőttek és gyermekek számára rutinszerű védőoltások javasoltak. Az antitesttitereket a vakcinázás után kell meghatározni, mivel a betegek immunogén válasza szuboptimális lehet. Szükség lehet a véralvadási faktorok profilaktikus pótlására bármilyen sebészeti beavatkozás előtt, ha a kiinduláskor hiányosságok állnak fenn.
Klinikai genetikai vizsgálatot kell végezni a CDG örökletes vonatkozásainak megvitatása, valamint az orvosi otthon kialakítása érdekében ezen összetett betegek számára. Az orvosi otthon általában a biokémiai genetikai szolgálat, bár a genetikai, idegfejlődési vagy neurológiai osztályok is szolgáltak már ebben a minőségben, ha nem áll rendelkezésre dedikált biokémiai genetikai szolgálat. Gyakran van szükség gasztroenterológiai, hematológiai, endokrinológiai, táplálkozási támogatás, beszéd-, foglalkozás-, fiziko- és táplálásterápia, ortopédia és rehabilitációs orvoslás szakorvosi beutalóra.
Célzott terápiák és prognózis
A CDG típusok többségének kezelése néhány kivételtől eltekintve nagyrészt támogató jellegű. Az MPI-CDG az összes CDG közül a leghatékonyabban kezelhető. Az orális mannózt intracelluláris hexokinázok alakítják át mannóz-6-foszfáttá, így megkerülve az enzimatikus blokkot és előállítva a hiányos szubsztrátot. A mannózpótlás jellemzően napi 1 g/testsúlykilogrammnál kezdődik, napi 4-6 adagra elosztva. Míg a potenciálisan életveszélyes fehérjepazarló enteropátia különösen jól reagál a mannózkezelésre, addig az MPI-CDG-ben a májbetegség tovább progrediálhat. A klinikai tünetek gyorsan javulnak és a transzferrin CDT hónapok alatt normalizálódik, bár a májbetegség a kezelés hatására tovább progresszálódhat (45,80,81).
A mannóz vemhesség alatti pótlásával óvatosan kell bánni, mivel a mannóz adása vemhes hipomorf foszfomannóz-izomeráz egérmodellekben embrionális letalitáshoz és a kölykök vakságához vezetett (82). Ezenkívül az intravénás mannóz beadásához csökkent tudatállapot és görcsrohamok társultak, amelyek glükóz beadásával megszűntek (83).
A PMM2-CDG kezelése nagyrészt szupportív és a tüneteken alapul. Ugyanakkor a mannóz-1-foszfát szubsztrátpótló terápiával kapcsolatos közelgő klinikai vizsgálatok jelenleg kidolgozás alatt állnak.
A többi CDG esetében különböző orális egyszerű cukrokat vizsgáltak azzal a céllal, hogy elméletileg javítsák a hipoglikozilációt. Az SLC35C1-CDG esetében a fukózt, a PGM1-CDG és az SLC35A2-CDG esetében pedig a galaktózt próbálták ki, vegyes eredményekkel (84). A D-galaktóz 1,0-2,5 g/kg/nap (max. 50 gramm) mennyiségben bizonyítottan javítja a hipoglikémiát, a koagulopátiát és az endokrinopátiát PGM1-CDG-ben (85,86). A galaktóz szintén javítja az endokrinopátiát és a koagulopátiát TMEM165-CDG (87) és SLC39A8-CDG esetében. Jelentős klinikai javulásról számoltak be SLC39A8-CDG betegeknél is, akik 15-20 mg/kg/nap MnSO4-et kaptak (88). Jelenleg is folynak klinikai vizsgálatok az N-acetilmannozamin (ManNAc) hasznosságának vizsgálatára a GNE-CDG-ben (89), és számos preklinikai vizsgálat van folyamatban más CDG-vel kapcsolatban (90).
Az orvosi előrelépések ellenére a CDG-ben szenvedő gyermekek esetében az első életévben jelentős a halálozás többszervi elégtelenség vagy súlyos fertőzés miatt (91). A CDG-ben szenvedő csecsemőknél előfordulhat fulmináns többszervi betegség, kezelhetetlen görcsrohamok vagy súlyos hypoalbuminémia, amely anasarca felé halad. Egyes betegek reagálnak az agresszív diurézisre és albuminpótlásra, míg mások nem reagálnak a kezelésre. A nátrium-butirát bizonyítottan javítja a rohamok kontrollját CAD-CDG és PIGM-CDG esetén (92). A ketogén diéta szintén csökkentette a rohamok gyakoriságát a PIGA-CDG egyes eseteiben (93). A stroke-szerű epizódok során az intravénás hidratálás és a normális vércukorszint fenntartása hasznos lehet, amíg a mögöttes érrendszeri trombotikus vagy vérzéses etiológiát kizárják.
A genomszerkesztési technikák megjelenésével és a CDG diagnosztikai ernyője alá tartozó betegségek mechanizmusának jobb megértésével a célzott terápiás fejlesztés jövője továbbra is ígéretes.
Köszönet
Köszönjük Lynne Wolfe, ARNP és Donna Krasnewich, MD, PhD számára, hogy a Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789) keretében kapott klinikai fotókat rendelkezésünkre bocsátották. Szeretnénk továbbá köszönetet mondani Jenny Thies, MS, LGC-nek a genetikai tanácsadásban szerzett szakértelméért.
Finanszírozás: IJ Chang a National Institutes of Health T32GM007454 támogatását élvezi.
Footnote
Conflicts of Interest:
Tájékoztatott beleegyezés:
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a gyorsan bővülő betegségcsalád. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Hardy-Weinberg egyensúly hiánya a CDG-Ia (congenitalis glycosylation disorders of glycosylation type Ia) leggyakoribb PMM2 mutációjára vonatkozóan. European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG nomenklatúra: ideje változtatni! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. Mi újság a CDG-ben? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Congenital disorders of glycosylation. I. rész: A fehérjék N-glikozilációjának hibái. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Az N-hez kötött glikánok intracelluláris funkciói. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Klinikai, laboratóriumi és molekuláris leletek és hosszú távú követési adatok 96 francia PMM2-CDG (phosphomannomutase 2-congenitalis disorder of glycosylation) betegnél és az irodalom áttekintése 96 francia betegnél. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. A PMM2-CDG országos felmérése Olaszországban: az L32R mutációhoz társuló enyhe neurológiai variáns nagy gyakorisága. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG Dél-Afrikában: Öt új beteg genotípus-fenotípus leírása. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Different glycosyltransferases are differentially processed for secretion, dimerization, and autoglycosylation. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In: Mora-Montes H. Editor. Glycans: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Bőrmanifesztációk CDG-ben. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF rendszer veleszületett glikozilációs rendellenességben szenvedő gyermekeknél. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. A klinikai megjelenési formák széles spektruma a veleszületett glikozilációs zavaroknál I: 26 esetből álló sorozat. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertális növekedés az Ia típusú veleszületett glikozilációs zavarban (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N-glycan synthesis. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Congenitalis glycosylation disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–a nemrég felfedezett örökletes anyagcsere-betegség. Több szervi manifesztáció, incidencia 1/80 000, nehezen kezelhető. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Súlyos átmeneti myocardialis ischaemia okozta hypertrophiás cardiomyopathia egy Ia típusú veleszületett glikozilációs zavarban szenvedő betegnél. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Pericardialis és hasi folyadékfelhalmozódás az Ia típusú veleszületett glikozilációs rendellenességben. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Az Ia típusú veleszületett glikozilációs rendellenességben (CDG-Ia) szenvedő felnőttek klinikai jellemzői. Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Szénhidráthiányos glikoprotein szindróma. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. A veleszületett glikozilációs zavar klinikai és molekuláris jellemzői 1-es típusú szialotranszferrin mintázatú és különböző etnikai eredetű betegeknél. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modifikáció a potyvírus Plum pox vírus köpenyfehérjéjében fokozza a vírusfertőzést. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Skandináv CDG-Ia betegek: genotípus/fenotípus korreláció és a founder mutációk földrajzi eredete. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 francia felnőtt PMM2-kongenitális glikozilációs rendellenességben szenvedő beteg: a klasszikus gyermekkori fenotípus kimenetele és egy későn jelentkező fenotípus ábrázolása. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. A PMM2-CDG veleszületett glikozilációs rendellenesség retinális jellemzői. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): az R141H/F119L genotípus fenotípusos spektruma. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Pajzsmirigyműködés PMM2-CDG-ben: diagnosztikai megközelítés és javasolt kezelés. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenitalis disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Új rendellenességek a szénhidrát-anyagcserében: veleszületett glikozilációs rendellenességek és hatásuk az endokrin rendszerre. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogenity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Az 1-es típusú szénhidráthiányos glikoprotein szindróma endokrinológiája a születéstől a serdülőkorig. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonádfunkció és glikoprotein hormonok a szénhidráthiányos glikoprotein (CDG) szindrómában. Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. A foszfomannóz-izomeráz hiány klinikai spektruma, a CDG-Ib mannózkezelésének értékelésével. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Foszfomannóz-izomeráz-hiány és a mannózterápia. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: A szénhidráthiányos glikoprotein szindróma új, mannózzal kezelhető megnyilvánulása. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG6 defektív veleszületett glikozilációs rendellenesség. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: A termeléstől a szekrécióig. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparán-szulfát a csontváz fejlődésében, növekedésében és patológiájában: az örökletes multiplex exosztózisok esete. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozigóta p.R270C mutáció alapító hatással Larsen of Reunion Island szindrómát okoz. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Különféle nem gyulladásos mozgásszervi betegségek. Hiperfoszfatémiás familiáris tumoros kalcinózis (FGF23, GALNT3 és αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. A Galnt3 gén ablációja alacsony intakt fibroblaszt növekedési faktor 23 (Fgf23) koncentrációhoz és hyperfoszfatémiához vezet a megnövekedett Fgf23 expresszió ellenére. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Egy gangliozid bioszintetikus enzim, az ST3GAL5 mutációja só & bors szindrómát eredményez, egy megváltozott glikolipid és glikoprotein glikozilációval járó neurokután rendellenességet. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutations in B4GALNT1 (GM2 synthase) underlie a gangliozid bioszintézis egy új rendellenessége. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA mutációk korai kezdetű epilepsziás encephalopátiákat és megkülönböztető jegyeket okoznak. Neurology. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodelling protein, cause autosomal-recessive intellectual disability. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutations in PIGO, a GPI-anchor-synthesis pathway egyik tagja, cause hyperphosphatasia with mental retardation. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. A PIGT homozigóta mutációi által okozott GPI-horgony hiány okozta új értelmi fogyatékossági szindróma. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) okozta szomatikus mutációk a PIG-A génben. EMBO J 1994;13:110-7.
- Brodsky RA. Paroxysmalis éjszakai hemoglobinuria. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. A tömegspektrometria hatása a veleszületett glikozilációs rendellenességek diagnosztikájában. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Exonok és intronok eloszlása az emberi genomban. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a Policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. A kortárs etika áramlatai. A GINA, az ADA és a genetikai diszkrimináció a foglalkoztatásban. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Recurrent thrombo-embolia in a child with a congenitalis disorder of glycosylation (CDG) type Ib and treatment with mannose. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Májbetegség kialakulása a mannózkezelés ellenére két CDG-Ib-ben szenvedő betegnél. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplements induce embryonic letality and blindness in phosphomannose isomerase hypomorphic mice. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Rohamok és szédülés intravénás mannózkezelés alatt egy 1b típusú CDG-szindrómás betegnél (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Fukózpótlás a II-es típusú leukocita adhéziós hiányban. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Öröklött glikozil-foszfatidil-inozitol hiány: kezelhető CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Orális D-galaktóz kiegészítés PGM1-CDG-ben. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Galaktózpótlás TMEM165-CDG-vel rendelkező betegeknél megmenti a glikozilációs hibákat. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8-hiány: biokémiai korrekció és jelentős klinikai javulás mangánterápiával. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Orális monoszacharid terápiák a vese- és izomhyposzialiláció visszafordítására a GNE myopathia egérmodelljében. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG terápiák: From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Humán glikozilációs rendellenességek. Marino PA, szerkesztő. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogén diéta – A PIGA hiány miatt kialakuló korai epilepsziás encephalopathia újszerű kezelése. Brain Dev 2016;38:848-51.