- Überblick
- Epidemiologie
- Biochemische Klassifizierung und Nomenklatur
- Genetik
- Pathophysiologie
- Defekte der N-gebundenen Proteinglykosylierung
- O-gebundene Glykosylierungsdefekte und kombinierte N- und O-gebundene Glykosylierungsdefekte
- Lipidglykosylierung und GPI-Anker-Biosynthesedefekte
- Klinische Manifestationen
- N-gebundene Proteinglykosylierungsdefekte
- PMM2-CDG (CDG-Ia, PMM2-Mangel)
- MPI-CDG (CDG-Ib, Mannosephosphat-Isomerase-Mangel)
- ALG6-CDG (Glucosyltransferase 1-Mangel)
- O-gebundene Glykosylierungsdefekte und kombinierte N- und O-gebundene Glykosylierungsdefekte
- Defekte der Lipidglykosylierung und GPI-Anker-Biosynthese
- Diagnose
- N-verknüpfte Proteinglykosylierungsdefekte
- O-gebundene Glykosylierungsdefekte und kombinierte N- und O-gebundene Glykosylierungsdefekte
- Defekte der Lipidglykosylierung und GPI-Anker-Biosynthese
- Molekulare Analyse
- Behandlung
- Gezielte Therapien und Prognose
- Danksagung
- Fußnote
Überblick
Glykosylierung ist der Prozess des Anhängens von Zuckerresten an Proteine und Lipide in verschiedenen zellulären Abläufen. Angeborene Glykosylierungsstörungen (CDG) sind eine genetisch und klinisch heterogene Gruppe von über hundert Krankheiten, die durch Defekte in verschiedenen Schritten der Glykansynthese oder -modifikation verursacht werden. Die meisten dieser monogenen Krankheiten werden autosomal rezessiv vererbt, aber es wurden auch autosomal dominante und X-chromosomale Formen beschrieben.
CDG zeigen typischerweise multisystemische Manifestationen, am häufigsten Entwicklungsverzögerung, Gedeihstörung, Hypotonie, neurologische Anomalien, Hepatopathie und Koagulopathie. Betroffene Personen können auch Augen-, Haut- und Herzerkrankungen sowie Gesichtsdysmorphien aufweisen. Obwohl bei der Mehrheit der Betroffenen neurologische Veränderungen und kognitive Verzögerungen zu beobachten sind, gibt es auch Fälle und sogar Typen, die keine neurologischen Manifestationen aufweisen. Angesichts der breit gefächerten klinischen und genetischen Ätiologie der CDG beruht die klinische Diagnose auf einem hohen Verdachtsindex bei einer multisystemischen Erkrankung.
Die Analyse von Kohlenhydratdefizit-Transferrin (CDT) im Serum ist der Screening-Test der ersten Wahl bei Patienten mit Verdacht auf CDG, ist jedoch auf N-Glykosylierungsdefekte mit Sialinsäuremangel beschränkt. Zu den weiterführenden Tests gehören die Analyse von dolicholverknüpften Glykanen und Gentests. Eine frühzeitige Diagnose dieser Gruppe exponentiell wachsender Krankheiten ist wichtig, da einige CDGs behandelbar sind. Die Behandlung von Glykosylierungsdefekten ist hauptsächlich unterstützend, obwohl es gezielte Therapien für MPI-CDG, SLC35C1-CDG, PIGM-CDG und PGM1-CDG gibt. Einzelheiten zu diesen Behandlungen finden Sie im Abschnitt „Zielgerichtete Therapien und Prognose“ weiter unten. Der Schwerpunkt dieser Übersichtsarbeit liegt auf den häufigsten Arten von CDG mit erkennbaren Phänotypen oder Behandlungen, wobei die Zielgruppe die Hausärzte sind.
Epidemiologie
Die Inzidenz und Prävalenz aller Arten von CDG insgesamt sind nicht gut ermittelt worden, obwohl weltweit über Patienten mit fast jedem ethnischen Hintergrund berichtet wurde und beide Geschlechter gleichermaßen betroffen sind. Die geschätzte Prävalenz in der europäischen und afroamerikanischen Bevölkerung liegt bei 1/10.000, basierend auf der Trägerhäufigkeit bekannter pathogener Varianten in 53 Genen (1-4). Die Prävalenz der am häufigsten diagnostizierten CDG, PMM2-CDG, liegt zwischen 1/20.000 in der niederländischen Bevölkerung und 1/77.000 in Estland, basierend auf einzelnen Berichten (5,6). Bis heute wurden für die meisten CDG-Typen weniger als 100 Fälle gemeldet.
Biochemische Klassifizierung und Nomenklatur
Gemeinsam werden CDG derzeit in vier Kategorien eingeteilt: (I) N-gebundene Glykosylierung, (II) O-gebundene Glykosylierung, (III) kombinierte N- und O-gebundene/mehrfache Glykosylierung und (IV) Defekte in der Biosynthese von Lipid- und Glykosylphosphatidylinositol-Ankern (GPI).
Der Defekt der N-gebundenen Proteinglykosylierung PMM2-CDG, früher bekannt als CDG Typ Ia, wurde 1980 von Jaeken als erster CDG beschrieben und ist bis heute der bei weitem häufigste CDG (7). PMM2-CDG wurde ursprünglich als „Kohlenhydratmangel-Glykoprotein-Syndrom“ bezeichnet, da bei den betroffenen Personen durch isoelektrische Fokussierung von Serumtransferrin multiple Glykoprotein-Anomalien festgestellt wurden. In der Vergangenheit wurden CDG nach Mustern der Transferrin-Isoform-Analyse klassifiziert – Typ-I-Muster wurden auf Defekte beim Zusammenbau und Transfer von dolicholgebundenen Glykanen zurückgeführt, die im Zytoplasma oder im ER lokalisiert sind, und Typ-II-Muster wurden auf Verarbeitungsdefekte im Golgi-Apparat zurückgeführt. Mit dem Aufkommen einer weit verbreiteten molekularen Diagnostik wurde die CDG-Nomenklatur 2008 aktualisiert, um die molekulare Ätiologie von Krankheiten zu spezifizieren, was das exponentielle Wachstum von Krankheitsverläufen und Störungen widerspiegelt, die nicht in die zuvor etablierten dichotomen Kategorien passen. Gegenwärtig wird die CDG-Nomenklatur durch den Namen des betroffenen Gens (nicht kursiv, Gennamen unter www.genenames.org), gefolgt von -CDG (z. B. PMM2-CDG), angegeben (8).
Genetik
Die überwiegende Mehrheit der angeborenen Glykosylierungsstörungen wird autosomal rezessiv vererbt, wobei eine Mutation von jedem asymptomatischen (Träger-)Elternteil vererbt wird. Um eine genetische Diagnose zu stellen, sind molekulare Tests, in der Regel mit Sequenzierungsmethoden der nächsten Generation, erforderlich. Ein elterlicher Test auf die bekannte Variante kann die Vererbung bzw. das de novo Auftreten bestätigen. Bei autosomal rezessivem Erbgang beträgt das Wiederholungsrisiko für Geschwister und jede Schwangerschaft eines betroffenen Individuums 25 %, wenn es betroffen ist, 50 %, wenn es ein asymptomatischer Träger ist, und 25 %, wenn es nicht betroffen ist.
Eine Handvoll CDG wird autosomal dominant vererbt (N-gebunden: GANAB-CDG, PRKCSH-CDG; O-verknüpft: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Weniger sind X-gebunden (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). Die meisten dominanten und einige X-chromosomale Formen von CDG sind auf De-novo-Mutationen zurückzuführen. Spezifische Krankheiten und Gene werden weiter unten im Abschnitt „Pathophysiologie“ beschrieben.
Mutationsdaten für alle veröffentlichten Gene für CDG sind in der Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) verfügbar. Informationen über spezifische Genvarianten sind in der Leiden Open Variation Database mit integrierten In-silico-Pathogenitätstools (http://www.lovd.nl/3.0/home) verfügbar. Klinische Zusammenfassungen zu bestimmten Genen finden sich in der Online Mendelian Inheritance in Man (http://www.omim.org/) oder in einem begrenzteren Umfang bei GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Angesichts der geringen Anzahl betroffener Patienten für die meisten CDG-Subtypen ist es schwierig, eine Genotyp-Phänotyp-Korrelation herzustellen.
Pathophysiologie
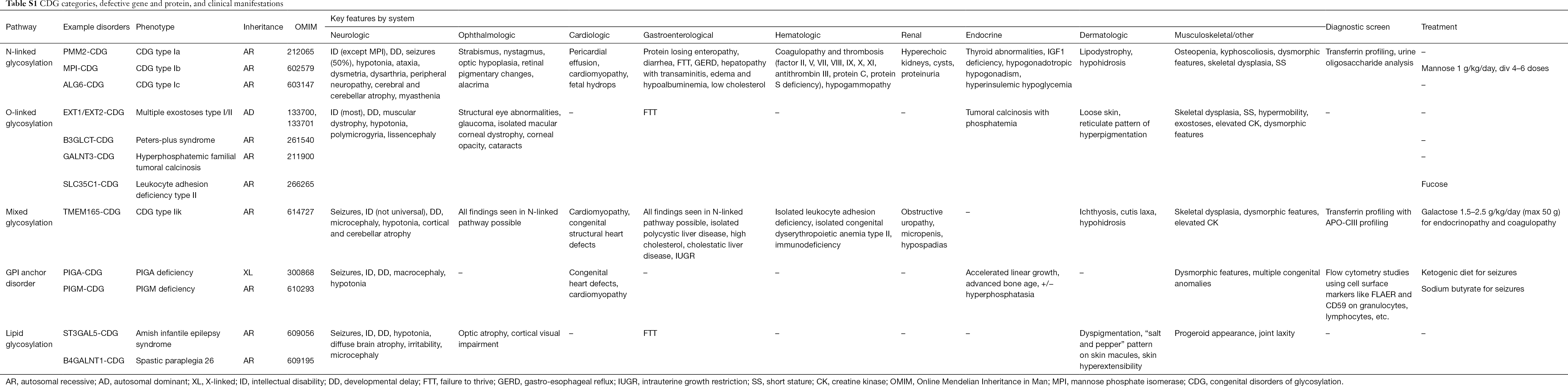
Bis heute sind über 130 Arten von CDG bekannt (9,10). Angesichts der ubiquitären Präsenz von Glykosylierungswegen sind CDG in ihrer biochemischen Pathogenese äußerst vielfältig. Zahlreiche Proteine und Lipide (d. h. Sphingolipide und Glykolipide) werden in verschiedenen zellulären Kompartimenten mit Monosacchariden und/oder Oligosacchariden, den so genannten Glykanen, glykosyliert. Ihre subzellulären Orte sind vielfältig, aber die meisten Defekte treten im ER oder Golgi-Apparat auf. Die klinischen Merkmale und die genetische Ätiologie der häufigsten CDG sind in Tabelle S1 zusammengefasst.
Unter den Proteinen werden die Glykane durch ihre Bindung an die Polypeptidkette beschrieben – N-Glykane sind an die Amidgruppe von Asparagin (Asn) gebunden, während O-Glykane an die Hydroxylgruppe von Serin oder Threonin gebunden sind. Die Synthese von N-Glykanen erfordert den schrittweisen Aufbau von nukleotidverknüpften Zuckern im Zytosol, den Zusammenbau im endoplasmatischen Retikulum und die Verarbeitung im Golgi-Apparat. Im Gegensatz dazu erfordert die O-Glykansynthese einen Zusammenbau, aber keine Verarbeitung, weshalb O-Glykosylierungsdefekte vorwiegend im Golgi-Apparat auftreten.
Defekte der N-gebundenen Proteinglykosylierung
N-Glykosylierung beinhaltet die kovalente Bindung von Kohlenhydratstrukturen an die Seitenketten-Amidgruppe von Asn-Resten innerhalb einer Konsensus-Asn-X-Ser/Thr-Akzeptorstelle, die Verlagerung des Substratpolypeptids zum endoplasmatischen Retikulum für den Umbau und die weitere Modifikation der N-Glykan-Kette im Golgi (11,12).
PMM2-CDG wird durch pathogene Varianten im Phosphomannomutase 2 (PMM2)-Gen verursacht, die zu einem Mangel des PMM2-Enzyms führen, das die zytosolische Umwandlung von Mannose-6-Phosphat in Mannose-1-Phosphat im zweiten Schritt der Guanosindiphosphat (GDP)-Mannosesynthese katalysiert. Bei den meisten Patienten liegen heterozygote pathogene Fehlmutationen vor (www.lovd.nl/PMM2). Die häufigste wiederkehrende pathogene Variante p.Arg141His findet sich bei etwa 40 % der betroffenen Personen europäischer Abstammung, und p.Phe119Leu wird ebenfalls häufig in Nordeuropa gefunden (1). Für PMM2-CDG wurden Genotyp-Phänotyp-Korrelationen berichtet (3, 13, 14).
MPI-CDG ist eine autosomal rezessive Erkrankung, die durch pathogene Varianten im Mannosephosphat-Isomerase (MPI)-Gen verursacht wird und zu einem Mangel an Phosphomannose-Isomerase (MPI) führt. MPI katalysiert normalerweise den ersten Schritt der GDP-Mannose-Synthese (d.h. die Umwandlung von Fructose-6-Phosphat in Mannose-6-Phosphat), aber Fructose-6-Phosphat sammelt sich nicht intrazellulär an, da es auch über den glykolytischen Weg verstoffwechselt werden kann. Daher verursacht MPI-CDG, obwohl biochemisch ähnlich wie PMM2-CDG, keine so signifikante neurologische und multisystemische Beteiligung. Der CDT ist auch der Screening-Test der Wahl für MPI-CDG, der ein Typ-1-Muster zeigt. Die Diagnose kann dann auf molekularer Ebene oder durch die MPI-Aktivität von Fibroblasten/Leukozyten bestätigt werden.
ALG6-CDG ist eine rezessive Erkrankung, die durch Mutationen in ALG6 verursacht wird und zu einer abnormalen Bindung von drei Glukosemolekülen an Dolichol-verknüpfte Mannose-Intermediate und einer nachgeschalteten Hypoglykosylierung von Serumglykoproteinen führt (15).
O-gebundene Glykosylierungsdefekte und kombinierte N- und O-gebundene Glykosylierungsdefekte
O-Glykosylierung umfasst die schrittweise Anlagerung von Kohlenhydratketten an Serin-, Threonin- und Hydroxylysinreste von Proteinen durch Glykosyltransferasen im Golgi-Apparat (16). Mehrere Arten von O-verknüpften Glykanen wurden mit menschlichen Krankheiten in Verbindung gebracht, benannt nach dem ersten Zucker, der an den Aminosäurerest gebunden ist (17).
Lipidglykosylierung und GPI-Anker-Biosynthesedefekte
GPI-Anker sind Glykolipide, die nacheinander im endoplasmatischen Retikulum aufgebaut und im Golgi modifiziert werden. GPI-Anker-Biosynthesestörungen, die auf Enzymmängel zurückzuführen sind, werden alphabetisch nach der Reihenfolge ihrer Entdeckung und nicht chronologisch nach dem Schritt des Zusammenbaus benannt. Sobald sie synthetisiert sind, befinden sich GPI-Anker auf Plasmamembranen und binden Hunderte von Zelloberflächenproteinen, die eine Vielzahl von Zellfunktionen erfüllen. Die meisten dieser Krankheiten werden autosomal rezessiv vererbt, mit der bemerkenswerten Ausnahme des X-chromosomalen PIGA-Mangels.
Klinische Manifestationen
Aufgrund der ubiquitären Präsenz von Glykosylierungswegen kann praktisch jedes Organsystem von CDG betroffen sein, obwohl die meisten Fälle neurologische Anomalien beinhalten. Einige CDG treten mit Ichthyose auf, darunter MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (Abbildung 1) und PIGL-CDG (18,19). Bei fast allen CDG treten in den ersten Lebensjahren multisystemische Erkrankungen auf, einige betreffen jedoch nur ein einziges Organsystem (z. B, Netzhaut bei DHDDS-CDG; neuromuskuläre Verbindung bei ALG2-CDG, ALG14-CDG, CFPT1-CDG; Gehirn bei ST3GAL3-CDG, TUSC3-CDG; Haut oder Skelettmuskel bei POGLUT1-CDG, POFUT1-CDG; Knorpel bei EXT1/EXT2-CDG; Leber bei TMEM199-CDG; rote Blutkörperchen bei SEC23B-CDG). Das Alter des Ausbruchs und der Schweregrad der Erkrankung können von der neonatalen Letalität bis zum nahezu asymptomatischen Erwachsenenalter und allen dazwischen liegenden Permutationen reichen. Zu den am häufigsten berichteten Symptomen gehören Entwicklungsverzögerung, Gedeihstörung, Hypotonie, neurologische Anomalien, Hypoglykämie und variable Leber-, Augen-, Haut-, Magen-Darm-, Immunologie-, Skelett- und Gerinnungsanomalien (19).
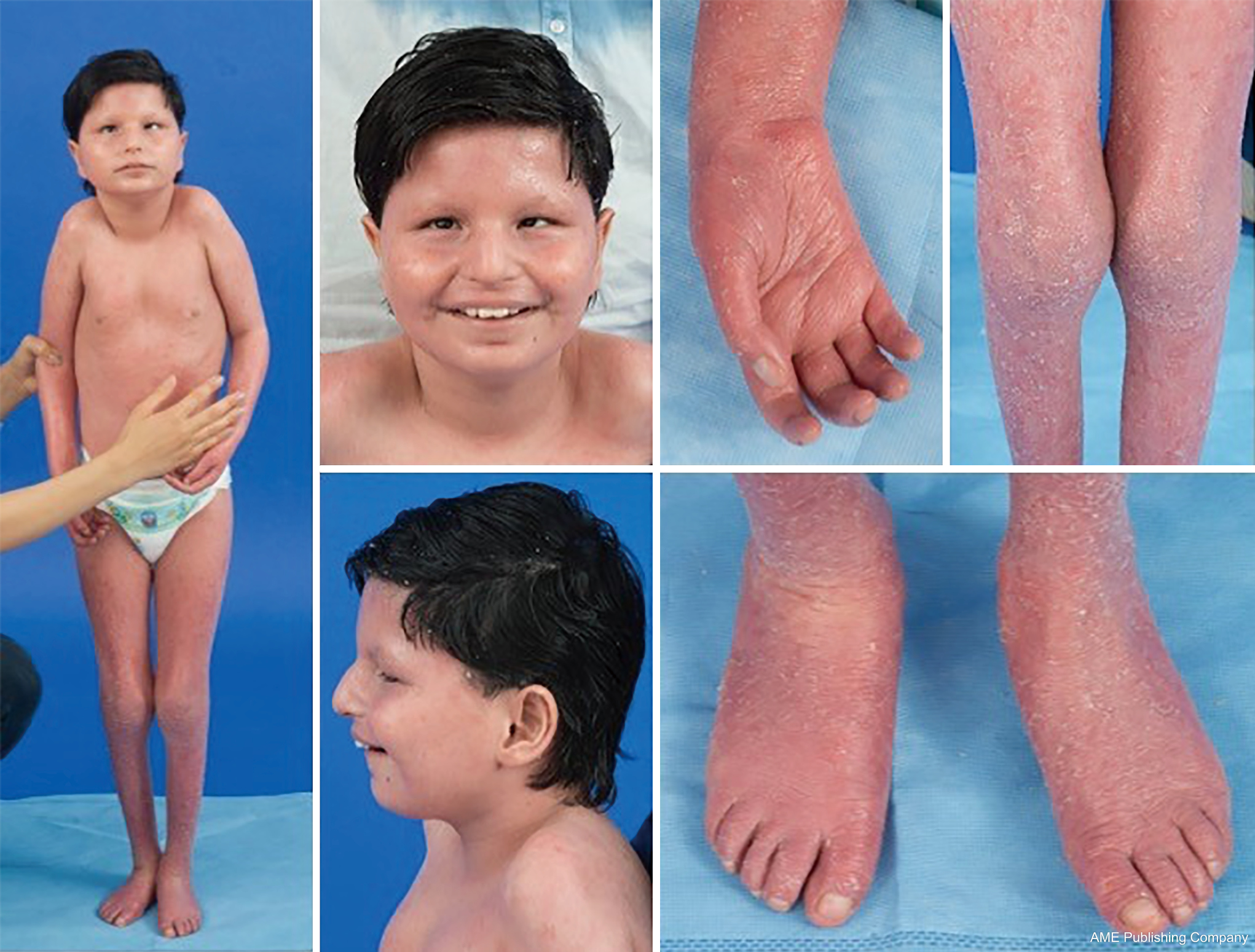
Der vollständige Phänotyp für viele CDG-Subtypen ist aufgrund der Seltenheit der gemeldeten Fälle noch nicht vollständig geklärt. Daher sollte CDG bei jeder multisystemischen Erkrankung in Betracht gezogen werden, insbesondere bei Fällen mit einer neurologischen Komponente oder einer unspezifischen Entwicklungsverzögerung mit unklarer Ätiologie.
Obwohl die Pathophysiologie einer Vielzahl von Symptomen noch geklärt werden muss, ist die Beziehung zwischen bestimmten Glykosylierungswegen und spezifischen klinischen Symptomen geklärt worden. So ist die Gedeihstörung, die bei vielen Arten von CDG auftritt, auf die Hypoglykosylierung und gestörte Bildung mehrerer Glykoproteine innerhalb des Insulin-Wachstumswegs zurückzuführen, darunter IGF-1, ALS und IGFBP-3 (20). In dem Maße, wie wir diese Gruppe komplexer Erkrankungen besser verstehen, werden CDG zunehmend bei Personen mit schwer fassbaren Diagnosen erkannt. Die Organsysteme, an denen die verschiedenen CDG beteiligt sind, sind in Tabelle S1 zusammengefasst. Nachfolgend werden die klinischen Merkmale der häufigsten Formen und der Formen mit gezielten Behandlungen von CDG erörtert.
Vollständige Tabelle
N-gebundene Proteinglykosylierungsdefekte
Als die am häufigsten diagnostizierten CDG wird der Phänotyp der N-gebundenen Glykosylierungsstörungen oft als das klassische Erscheinungsbild angepriesen. Das phänotypische Spektrum der CDG ist jedoch sehr vielfältig, und viele CDG zeigen nicht die stereotypen Symptome, die mit PMM2-CDG assoziiert sind.
PMM2-CDG (CDG-Ia, PMM2-Mangel)
PMM2-CDG ist mit über 700 gemeldeten Fällen weltweit die häufigste CDG. Sie ist gekennzeichnet durch eine schwere multisystemische Erkrankung im Säuglingsalter, eine neurologische Erkrankung und Entwicklungsverzögerung in der Kindheit und/oder eine stabile geistige Behinderung im Erwachsenenalter (21,22).
Im Säuglingsalter zeigt PMM2-CDG neurologische Anomalien, die typischerweise kurz nach der Geburt auftreten, nämlich Strabismus und abnorme Augenbewegungen, Kleinhirnhypoplasie, Hypotonie, psychomotorische Retardierung, Ataxie, Hypotonie und Hyporeflexie. Bei Säuglingen können auch Lebererkrankungen, nephrotisches Syndrom und Nierenzysten, Herzbeutelerguss und hypertrophe Kardiomyopathie, Gedeihstörungen und Multiorganversagen auftreten, die bei bis zu 20 % der Betroffenen innerhalb des ersten Lebensjahres zum Tod führen (21, 23-28).
Bei Patienten mit PMM2-CDG wurde eine Konstellation dysmorpher Merkmale beschrieben (Abbildungen 2, 3). Dazu gehören ein hypoplastisches Kleinhirn, Gesichtsdysmorphien (d. h. große, dysplastische Ohren), invertierte Brustwarzen und eine abnorme Verteilung des Fettgewebes über dem Gesäß oder der suprapubischen Region, die sich mit zunehmendem Alter zurückbilden kann (14, 21, 29-32). Die Patienten werden als aufgeschlossen und fröhlich beschrieben. Das Erscheinungsbild ist sehr unterschiedlich, obwohl bei mehr als 70 % der betroffenen Patienten ein Strabismus zu beobachten ist (21,23,33-35). Umgekehrte Brustwarzen und abnorme Fettpolster werden bei etwa 25-50 % der Patienten beobachtet (36).

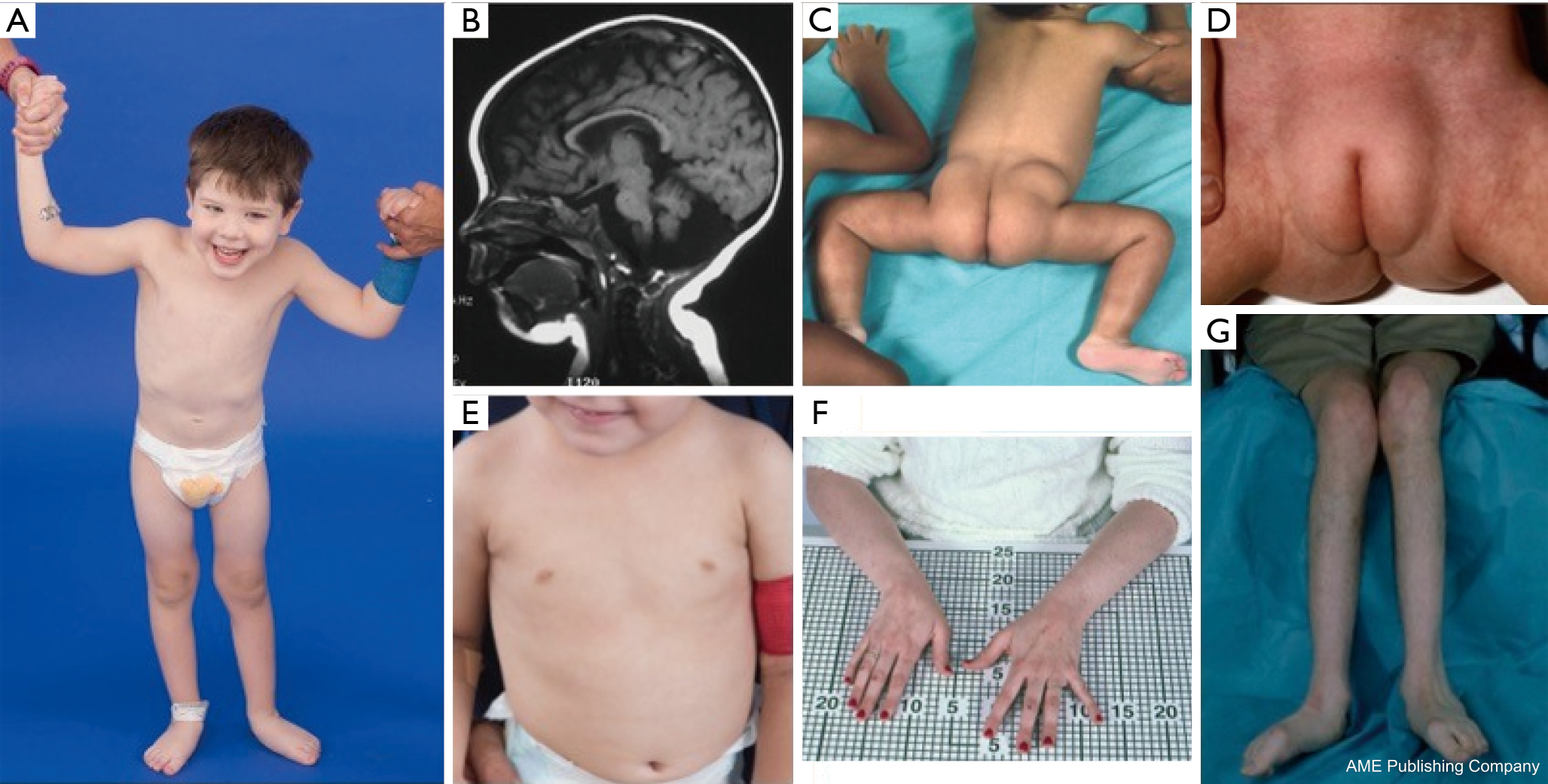
Im Kindesalter können die Betroffenen Retinitis pigmentosa, schlaganfallähnliche Episoden und Krampfanfälle, Sprach- und Bewegungsverzögerungen sowie periphere Neuropathie entwickeln. Konstitutionell haben die Patienten häufig Gedeihstörungen aufgrund von Fütterungs- und Magen-Darm-Anomalien sowie eine globale Entwicklungsverzögerung. Erhöhte Lebertransaminasen ohne klinische Folgen können beobachtet werden, die sich in der Regel bis zum Alter von 5 Jahren normalisieren, wobei es gelegentlich zu krankheitsbedingten Schwankungen kommt (21, 24). Leberbiopsien sind bei CDG nur selten angezeigt, es sei denn, es besteht der Verdacht auf eine Leberfibrose (1). Eine klinische Hypothyreose ist selten, aber bei Patienten mit CDG sollten die Schilddrüsenhormone und das freie T4 gemessen werden, was einen niedrigen Wert des schilddrüsenbindenden Globulins (TBG) und vorübergehende Erhöhungen des schilddrüsenstimulierenden Hormons (TSH) ergeben kann (37). Über Fehlbildungen der Leber und der Gallenwege wurde bei PMM2-CDG-Patienten nicht berichtet.
Erwachsene mit PMM2-CDG können bis zum 7. oder 8. Lebensjahrzehnt mit stabiler kognitiver Verzögerung, peripherer Neuropathie und progressiver thorakaler und spinaler Kyphoskoliose mit Osteopenie oder Osteoporose leben (34). Die zerebelläre Ataxie ist ein zunehmend anerkanntes Symptom zusammen mit einer multisystemischen Beteiligung (38-40). Endokrine Anomalien wie Hyperprolaktinämie, Wachstumshormonausschüttung mit Hyperglykämie, Insulinresistenz und hyperinsulinämische Hypoglykämie (41,42). Bei den betroffenen Frauen kann der hypogonadotrope Hypogonadismus zu einer fehlenden sekundären Geschlechtsentwicklung oder fehlenden Eierstöcken führen (41,43,44). Die Patienten können aufgrund verminderter Serumgerinnungsfaktoren, einschließlich der Faktoren IV, IX und XI, Antithrombin III, Protein C und Protein S, einem erhöhten Thromboserisiko ausgesetzt sein (29).
MPI-CDG (CDG-Ib, Mannosephosphat-Isomerase-Mangel)
MPI-CDG ist einzigartig, da die betroffenen Patienten nur wenig bis gar keine neurologische Beteiligung aufweisen und einige Manifestationen der Krankheit durch orale Mannose behandelbar sind (2). Die Symptome sind hauptsächlich hepatisch-intestinal, ohne dysmorphe Merkmale oder kognitive Verzögerungen. Die Patienten weisen typischerweise rezidivierendes Erbrechen, signifikante Hypoglykämie, Gedeihstörungen, eine potenziell lebensbedrohliche Proteinverlust-Enteropathie, fibrotische Leberveränderungen und eine Gallengangserweiterung auf (45-51). Die Patienten haben ein erhöhtes Risiko für thrombotische Ereignisse aufgrund niedriger Serumkonzentrationen von Protein C und S und Anti-Thrombin III.
ALG6-CDG (Glucosyltransferase 1-Mangel)
ALG6-CDG ist der zweithäufigste N-Glykosylierungsdefekt, der durch einen ähnlichen, aber milderen Phänotyp als PMM2-CDG gekennzeichnet ist. Patienten mit ALG6-CDG haben Gedeihstörungen, Entwicklungsverzögerungen, Hypotonie, Krampfanfälle, Strabismus, Ataxie, Koagulopathie und Gesichtsdysmorphien (d. h. tief angesetzte Ohren, Hypertelorismus und Makroglossie). Ähnlich wie bei MPI-CDG können sie auch eine Proteinverlust-Enteropathie haben. Außerdem können die betroffenen Patienten Skelettanomalien wie Brachydaktylie, Fingerfehlbildungen und Skoliose aufweisen. Betroffene Patienten haben typischerweise keine Retinitis pigmentosa oder Kleinhirnhypoplasie (52).
O-gebundene Glykosylierungsdefekte und kombinierte N- und O-gebundene Glykosylierungsdefekte
Da O-Glykane in mucinhaltigen Proteinen, einschließlich Glykosaminoglykanen (GAGs) und Epitheloberflächen, stark vertreten sind (53), führen Störungen der GAG-Synthese typischerweise zu Skelettdysplasien oder Bindegewebserkrankungen. Betroffene Patienten können neben neurologischen Symptomen auch Anomalien des Muskel-Skeletts, der Haut und der Gelenke aufweisen (z. B. Gelenklaxität, multiple Exostosen, Chondro-/Osteosarkome) (54-56). Die N-Acetylgalactosaminyltransferase 3 (GALNT3) beispielsweise O-glykosyliert das Phosphatbildungshormon FGF23, verhindert so die proteolytische Spaltung und ermöglicht dessen intakte Sekretion. Ein GALNT3-Mangel führt zu einer familiären tumoralen Kalzinose, die durch Hyperphosphatämie und ektopische Verkalkungen gekennzeichnet ist (57,58).
Defekte der Lipidglykosylierung und GPI-Anker-Biosynthese
Glykosphingolipide und ihre sialylierten Derivate, Ganglioside, werden hauptsächlich von Neuronen exprimiert. Defekte beim Abbau von Gangliosiden führen zur Akkumulation und zu den gut charakterisierten lysosomalen Speicherkrankheiten. Auf der anderen Seite sind Defekte in der Gangliosid-Biosynthese, wie ST3GAL5-CDG und B4GALNT1-CDG, äußerst selten und führen zu schweren neurodegenerativen Erkrankungen. Die Patienten können spastische Paraplegie, schwere intellektuelle Verzögerung, Epilepsie und nicht-neurologische Symptome wie Skelettdysplasie, dysmorphe Merkmale und abnormale Hautpigmentierung aufweisen (59,60).
Mutationen in vielen Genen innerhalb des GPI-Anker-Biosynthesewegs verursachen eine Vielzahl von kongenitalen Anomalien, intellektuelle Behinderung und Epilepsie. Der am besten charakterisierte GPI-Biosynthesedefekt, der X-chromosomale PIGA-Mangel, äußert sich in kindlichen Krämpfen mit Hypsarrhythmie, Hypotonie, multiplen Gehirnanomalien und Gesichtsdysmorphien. Die Patienten können auch unterschiedliche Haut-, Leber-, Herz- und Nierenerkrankungen haben (61-69). Einige Mutationen innerhalb von PIGA verursachen die phänotypisch unterschiedliche Krankheit paroxysmale nächtliche Hämoglobinurie (PNH), eine erworbene Störung des Knochenmarkversagens (70,71).
Diagnose
Wenn ein CDG klinisch vermutet wird, besteht der erste Schritt darin, biochemische CDG-Tests in Plasma oder Serum anzuordnen, einschließlich CDT- und N-Glykan-Tests. CDT- und N-Glykan-Analysen im Serum können nur N-Glykosylierungsdefekte nachweisen und sind daher für die Unterscheidung von isolierten O-Glykosylierungs- oder GPI-Ankerdefekten nicht geeignet. Die Analyse der Transferrin-Isoformen erfolgte ursprünglich durch isoelektrische Fokussierung von Transferrin, da ein Ausfall der N-Glykansynthese einen teilweisen Mangel an Sialinsäure verursacht, der die Ladung des Serumtransferrins und damit seine kathodische Wanderung auf einem elektrophoretischen Feld verändert. Die massenspektrometrische Analyse von Transferrin und N-Glykan hat die isoelektrische Fokussierung jedoch inzwischen weitgehend ersetzt, indem sie spezifische Veränderungen der Oligosaccharide durch Masse und Ladung identifiziert (72).
N-verknüpfte Proteinglykosylierungsdefekte
Serumtransferrin CDT-Ergebnisse werden als das Verhältnis von Mono-Oligosaccharid/Di-Oligosaccharid-Transferrin angegeben, a-Oligosaccharid/Di-Oligosaccharid-Transferrin, Tri-Sialo/Di-Oligosaccharid-Transferrin, Apolipoprotein CIII-1/Apolipoprotein CIII-2 und Apolipoprotein CIII-0/Apolipoprotein CIII-2 Verhältnis. Zu diesen quantitativen Ergebnissen gehört auch eine Interpretation des Befundmusters.
Ein Transferrin-CDT-Muster vom Typ I ist durch erhöhte Di- und Asialotransferrinbanden gekennzeichnet und weist auf Defekte in der N-Glykansynthese im Zytosol oder im endoplasmatischen Retikulum hin. Ein Typ-II-Muster ist durch erhöhte Di- und Asialotransferrinbanden sowie Tri- und/oder Monosialotransferrinbanden gekennzeichnet und deutet auf Defekte bei der N-Glykanverarbeitung im Golgi-Apparat hin (73).
Wird ein Serum-Transferrin-CDT-Muster vom Typ I festgestellt, sollten PMM2-Mangel oder MPI-Mangel im Vordergrund der Differenzialdiagnose stehen, da PMM2-CDG die häufigste CDG ist und MPI-CDG behandelbar und unbehandelt potenziell tödlich ist. Zur Differenzierung zwischen den Diagnosen sollten N-Glykan-Profilierung, molekulare Sequenzierung oder enzymatische Tests durchgeführt werden. Die Diagnose von PMM2-CDG oder MPI-CDG wird durch molekulare Tests bestätigt, die biallelische pathogene Varianten in PMM2 oder MPI zeigen, gefolgt von PMM- oder MPI-Enzymaktivität in Leukozyten oder Fibroblasten, wenn die Pathogenität der genetischen Varianten unklar ist. Eine N-Glykan-Analyse oder molekulare Analyse würde die meisten ALG-CDG von PMM2- oder MPI-CDG unterscheiden (15).
Ein Serum-Transferrin-CDT-Muster vom Typ II deutet auf Golgi-Defekte wie N-Acetylglucosaminyltransferase (GnT) II-Mangel hin (CDG Typ IIA, MGAT2-CDG). Die Analyse der Isoform von Apolipoprotein CIII (Apo-CIII) ist ein ergänzender Test für ein CDT-Profil vom Typ II, da sie Defekte der Mucin-O-Glykosylierung im Golgi-Apparat misst. Die Empfindlichkeit von CDT oder Apo-CIII beim Nachweis von CDG Typ II ist begrenzt. Daher sollten N-Glykan- und O-Glykan-Profile und molekulare Panel- oder Exom-Sequenzierungen durchgeführt werden, wenn diese klinischen Tests verfügbar sind. Transferrin-Glykosylierungsmuster können sich sporadisch normalisieren; daher können wiederholte Tests bei Patienten mit hohem Verdachtsindex angezeigt sein. Falsch positive Ergebnisse können bei Patienten mit einer akuten Krise der hereditären Fruktoseintoleranz, Galaktosämie, akuten Lebererkrankungen und einigen bakteriellen Infektionen auftreten. Keiner der biochemischen CDG-Tests kann auf alle CDGs testen, so dass selbst bei normalen Screening-Ergebnissen bei starkem klinischen Verdacht ein molekularer Gentest oder eine Exom-Sequenzierung durchgeführt werden kann. Umgekehrt sind biochemische und funktionelle Bestätigungen molekulargenetischer Befunde ebenfalls unerlässlich, da die Mehrheit der Patienten mit CDG mindestens eine leichte und oft neuartige Missense-Mutation trägt.
O-gebundene Glykosylierungsdefekte und kombinierte N- und O-gebundene Glykosylierungsdefekte
Die Diagnose hängt von der molekularen Sequenzierung ab, da eine Transferrin-Isoform-Analyse isolierte O-Glykosylierungsdefekte nicht erkennen würde. Kombinierte N- und O-gebundene Glykosylierungsdefekte können durch CDT, ApoCIII-Analyse und Plasma-N-Glykan- und O-Glykan-Analyse nachgewiesen werden.
Defekte der Lipidglykosylierung und GPI-Anker-Biosynthese
Die Durchflusszytometrie von Blutgranulozyten misst die Zelloberflächenexpression von GPI-verankerten Proteinen wie CD16 und CD24. Die Durchflusszytometrie-Analyse von weißen oder roten Blutkörperchen auf bestimmte GPI-verankerte Zelloberflächenproteine steht klinisch als Test für PNH aufgrund erworbener Mutationen im PIGA-Gen zur Verfügung. Der PNH-Test kann Anomalien bei anderen GPI-Anker-Mängeln aufdecken, aber die Diagnose beruht meist auf einer molekularen Analyse.
Molekulare Analyse
Die höchste diagnostische Ausbeute für CDG ist ein Gen-Sequenzierungs-Panel der nächsten Generation oder die klinische Exom-Sequenzierung (CES). Bei der genetischen Sequenzierung wird die Nukleotidsequenz oder „Buchstaben“-Schreibweise von Genen überprüft, um festzustellen, ob eine Veränderung vorliegt, die die Funktion des Gens beeinträchtigt. Das menschliche Genom besteht aus 3 Millionen Nukleotiden, aber nur 1-2 % davon, die so genannten Exons, werden in ein funktionelles Proteinprodukt übersetzt. Die verbleibende nicht codierende DNA, die zwischen den Exons liegt und nicht übersetzt wird, wird als Introns bezeichnet (74). Bei der CES werden fast alle bekannten Exons der rund 20 000 Gene im menschlichen Genom untersucht, die nur eine Minderheit des genetischen Materials in den Chromosomen ausmachen, aber mit großer Wahrscheinlichkeit krankheitsverursachende (pathogene) Varianten enthalten. CES kann auch die Sequenzierung mitochondrialer DNA (mtDNA) umfassen, die kleine, außerkernige, zirkuläre DNA in den Mitochondrien abfragt, die ausschließlich mütterlicherseits vererbt wird.
Die möglichen Ergebnisse für CES umfassen positive, negative und Varianten unbekannter Bedeutung. Ein positives Ergebnis bedeutet, dass bekannte krankheitsverursachende (d. h. pathogene) Varianten identifiziert wurden, woraufhin die Diagnose, der natürliche Verlauf, die Prognose, das Rezidivrisiko und die Behandlungsmöglichkeiten diskutiert werden können. Ein negatives Ergebnis bedeutet, dass keine nachweisbaren pathogenen Varianten identifiziert wurden. Varianten unbekannter Signifikanz (VUS) bedeuten, dass zwar genetische Veränderungen identifiziert wurden, aber nicht genügend Informationen über die spezifische genetische Veränderung vorliegen, um definitiv zu wissen, ob sie krankheitsverursachend ist. Da Abweichungen in der DNA jedes Einzelnen zu erwarten sind, kann die gleichzeitige Untersuchung der elterlichen Proben zum Vergleich die Interpretation der Ergebnisse im Labor und in der Klinik erleichtern. Die diagnostische Ausbeute der CES wird auf 30-35 % geschätzt und nimmt im Laufe der Zeit zu, da die Entdeckung von Genen und das Wissen über das menschliche Genom weiter voranschreiten (75-77). Die CES wird zunehmend als erste Wahl für breit angelegte Gentests eingesetzt, da sie eine kurze Durchlaufzeit hat und im Verhältnis zur Menge der analysierten genetischen Informationen geringe Kosten verursacht. Zu den Einschränkungen der CES gehören die fehlende 100-prozentige Sensitivität, die Unfähigkeit, bestimmte Arten von genetischen Veränderungen (z. B. Deletionen, Duplikationen, Trinukleotid-Wiederholungen, tiefe intronische Mutationen oder Methylierungsdefekte) zu erkennen, und die Tatsache, dass eine Diagnose möglicherweise keine zusätzlichen Informationen über die Krankheit liefert oder das Management verändert.
Bei der Meldung von CES können zufällig entdeckte pathogene Varianten in Genen, die mit bekannten genetischen Erkrankungen assoziiert sind, als sekundäre Befunde gemeldet werden (78). Diese Liste der empfohlenen Krankheiten wurde vom American College of Medical Genetics (ACMG) zusammengestellt. Der Genetic Information Nondiscrimination Act (GINA) ist eine wichtige Überlegung bei der Entscheidung, ob man sich für oder gegen die Erfassung von Zufallsbefunden entscheidet (79). GINA schützt Einzelpersonen vor dem Missbrauch genetischer Informationen in der Krankenversicherung und bei der Beschäftigung, nicht aber bei Lebensversicherungen. GINA schützt die folgenden genetischen Informationen: Familienanamnese, Trägertests, pränatale Gentests, Anfälligkeits- und prädiktive Tests sowie die Analyse von Tumoren oder andere Bewertungen von Genen, Mutationen oder Chromosomenveränderungen.
Behandlung
Die Behandlung von CDG hängt weitgehend von den spezifischen Symptomen des Patienten ab. Zu den wiederkehrenden Symptomen bei Patienten mit CDG gehören Gedeihstörung, globale Entwicklungsverzögerung, Erbrechen, schlaganfallartige Anfälle und Skelettanomalien. Klinische oder subklinische Koagulopathie, Endokrinopathie, Hepatopathie und kardiale Defekte sind ebenfalls häufig zu beobachten. Vor allem bei PMM2-CDG werden Basis-Labortests zur Bestimmung des Krankheitsausmaßes und zur Routineüberwachung empfohlen. Dazu gehören Leberfunktionstests, Serumalbumin, Schilddrüsenfunktionstests einschließlich freiem T4, Protein C, Protein S, Antithrombin III, Faktor IX, Urinanalyse sowie Serumgonadotropine und Wachstumshormon.
Zu den empfohlenen bildgebenden Verfahren gehören Echokardiogramm, Nierenultraschall, Knochenalter, ophthalmologische Untersuchung zur Beurteilung von Linse, Netzhaut, Augenbeweglichkeit und Augeninnendruck. Sofern nicht anders angegeben, werden für Erwachsene und Kinder mit CDG Routineimpfungen empfohlen. Nach der Impfung sollten Antikörpertiter bestimmt werden, da die Patienten möglicherweise eine suboptimale Immunreaktion zeigen. Eine prophylaktische Auffüllung der Gerinnungsfaktoren vor chirurgischen Eingriffen kann erforderlich sein, wenn zu Beginn ein Mangel besteht.
Eine klinisch-genetische Untersuchung sollte durchgeführt werden, um die erblichen Aspekte der CDG zu erörtern und eine medizinische Anlaufstelle für diese komplexen Patienten zu finden. In der Regel handelt es sich dabei um die Abteilung für biochemische Genetik, aber auch Abteilungen für Genetik, Neuroentwicklung oder Neurologie haben diese Aufgabe übernommen, wenn eine spezielle Abteilung für biochemische Genetik nicht zur Verfügung steht. Eine Überweisung zum Facharzt für Gastroenterologie, Hämatologie, Endokrinologie, Ernährungsberatung, Sprach-, Ergo-, Physio- und Ernährungstherapie, Orthopädie und Rehabilitationsmedizin ist häufig erforderlich.
Gezielte Therapien und Prognose
Die Behandlung der meisten CDG-Typen ist weitgehend unterstützend, mit einigen Ausnahmen. MPI-CDG ist von allen CDG-Typen am wirksamsten behandelbar. Orale Mannose wird von intrazellulären Hexokinasen in Mannose-6-Phosphat umgewandelt, wodurch die enzymatische Blockade umgangen und das fehlende Substrat produziert wird. Die Mannose-Supplementierung beginnt in der Regel mit 1 g/kg Körpergewicht pro Tag, aufgeteilt in 4-6 Dosen pro Tag. Während die potenziell lebensbedrohliche proteinverzehrende Enteropathie besonders gut auf die Mannosebehandlung anspricht, kann die Lebererkrankung bei MPI-CDG weiter fortschreiten. Die klinischen Symptome bessern sich rasch und der Transferrin-CDT-Wert normalisiert sich im Laufe von Monaten, obwohl die Lebererkrankung unter der Behandlung weiter fortschreiten kann (45, 80, 81).
Bei der Verabreichung von Mannose während der Schwangerschaft ist Vorsicht geboten, da die Verabreichung von Mannose in schwangeren hypomorphen Phosphomannose-Isomerase-Mausmodellen zu embryonaler Letalität und Blindheit der Welpen führte (82). Darüber hinaus wurde die intravenöse Verabreichung von Mannose mit Bewusstseinsstörungen und Krampfanfällen in Verbindung gebracht, die sich mit der Verabreichung von Glukose auflösten (83).
Die Behandlung von PMM2-CDG ist weitgehend unterstützend und basiert auf der Symptomatik. Derzeit werden jedoch klinische Studien zur Mannose-1-Phosphat-Substrat-Ersatztherapie entwickelt.
Für andere CDG wurden verschiedene orale Einfachzucker mit dem Ziel untersucht, die Hypoglykosylierung theoretisch zu verbessern. Fucose wurde für SLC35C1-CDG und Galaktose für PGM1-CDG und SLC35A2-CDG mit gemischten Ergebnissen getestet (84). D-Galaktose in einer Dosierung von 1,0-2,5 g/kg/Tag (max. 50 g) verbessert nachweislich die Hypoglykämie, Koagulopathie und Endokrinopathie bei PGM1-CDG (85,86). Galaktose hat auch die Endokrinopathie und die Koagulopathie bei TMEM165-CDG (87) und SLC39A8-CDG verbessert. Auch bei SLC39A8-CDG-Patienten, die 15-20 mg/kg/Tag MnSO4 erhielten, wurde eine erhebliche klinische Verbesserung festgestellt (88). Derzeit laufen klinische Studien zur Untersuchung des Nutzens von N-Acetylmannosamin (ManNAc) bei GNE-CDG (89), und mehrere vorklinische Studien für andere CDG sind im Gange (90).
Trotz medizinischer Fortschritte besteht bei Kindern mit CDG innerhalb des ersten Lebensjahres eine erhebliche Sterblichkeit aufgrund von Multiorganversagen oder schweren Infektionen (91). Säuglinge mit CDG können eine fulminante Multiorganerkrankung, hartnäckige Krampfanfälle oder eine schwere Hypoalbuminämie aufweisen, die sich zu einer Anasarkose entwickelt. Einige Patienten sprechen auf eine aggressive Diurese und Albuminersatz an, während andere auf eine Behandlung nicht ansprechen. Natriumbutyrat verbessert nachweislich die Anfallskontrolle bei CAD-CDG und PIGM-CDG (92). Auch eine ketogene Diät hat in einigen Fällen von PIGA-CDG nachweislich die Anfallshäufigkeit verringert (93). Bei schlaganfallähnlichen Episoden kann eine intravenöse Flüssigkeitszufuhr und die Aufrechterhaltung eines normalen Blutzuckerspiegels hilfreich sein, während eine zugrunde liegende thrombotische oder blutende Ätiologie ausgeschlossen wird.
Mit dem Aufkommen von Genom-Editierungstechniken und einem besseren Verständnis der Mechanismen von Krankheiten, die unter den diagnostischen Oberbegriff CDG fallen, bleibt die Zukunft der gezielten Therapieentwicklung vielversprechend.
Danksagung
Wir danken Lynne Wolfe, ARNP, und Donna Krasnewich, MD, PhD, für die Bereitstellung von klinischen Fotos, die wir im Rahmen der Clinical and Basic Investigations Into Known and Suspected Congenital Disorders of Glycosylation (NCT02089789) erhalten haben. Wir möchten auch Jenny Thies, MS, LGC für ihre Expertise in der genetischen Beratung danken.
Finanzierung: IJ Chang wird von den National Institutes of Health T32GM007454 unterstützt.
Fußnote
Interessenkonflikte: Die Autoren haben keine Interessenkonflikte zu deklarieren.
Informierte Zustimmung: Die schriftliche Einwilligung der Patienten zur Veröffentlichung dieses Manuskripts und der zugehörigen Bilder wurde eingeholt.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutationen in PMM2, die zu kongenitalen Störungen der Glykosylierung, Typ Ia (CDG-Ia) führen. Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Fehlen eines Hardy-Weinberg-Gleichgewichts für die häufigste PMM2-Mutation bei CDG-Ia (congenital disorders of glycosylation type Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. CDG nomenclature: time for a change! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Congenital disorders of glycosylation. Part I. Defects of protein N-glycosylation. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Klinische, labortechnische und molekulare Befunde und Langzeit-Follow-up-Daten bei 96 französischen Patienten mit PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) und Überprüfung der Literatur. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG in South Africa: Genotype-Phenotype Description of Five Novel Patients. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Different glycosyltransferases are differentially processed for secretion, dimerization, and autoglycosylation. Glycobiology 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Congenital Disorders of Glycosylation. In: Mora-Montes H. Editor. Glycans: Biochemistry, Characterization and Applications Congenital Disorders of Glycosylation (Angeborene Störungen der Glykosylierung). Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Congenital disorders of glycosylation: other causes of ichthyosis. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Skin manifestations in CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. IGF system in children with congenital disorders of glycosylation. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Prepubertal growth in congenital disorder of glycosylation type Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Klinische und biochemische Merkmale der kongenitalen Glykosylierungsstörung Typ Ic, des ersten anerkannten Defekts des endoplasmatischen Retikulums bei der N-Glykan-Synthese. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–eine kürzlich entdeckte erbliche Stoffwechselerkrankung. Multiple Organmanifestationen, Inzidenz 1/80.000, schwierig zu behandeln. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Perikardiale und abdominale Flüssigkeitsansammlung bei angeborener Störung der Glykosylierung Typ Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Klinische Merkmale bei Erwachsenen mit kongenitalen Störungen der Glykosylierung Typ Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Carbohydrate-deficient glycoprotein syndrome. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Klinische und molekulare Merkmale einer angeborenen Glykosylierungsstörung bei Patienten mit Typ 1 Sialotransferrin-Muster und unterschiedlicher ethnischer Herkunft. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. O-GlcNAc modification of the coat protein of the potyvirus Plum pox virus enhances viral infection. Virology 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Scandinavian CDG-Ia patients: genotype/phenotype correlation and geographic origin of founder mutations. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 französische erwachsene Patienten mit PMM2-kongenitaler Glykosylierungsstörung: Ergebnis des klassischen pädiatrischen Phänotyps und Darstellung eines spät einsetzenden Phänotyps. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Thyroid function in PMM2-CDG: diagnostic approach and proposed management. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early-onset cerebellar ataxia in an adult. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Neue Störungen des Kohlenhydratstoffwechsels: angeborene Störungen der Glykosylierung und ihre Auswirkungen auf das endokrine System. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Gonadenfunktion und Glykoprotein-Hormone beim Kohlenhydrat-Mangel-Glykoprotein-Syndrom (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Kardiale Komplikationen bei kongenitalen Störungen der Glykosylierung (CDG): eine systematische Übersicht über die Literatur. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Rezidivierender nicht-immuner Hydrops fetalis assoziiert mit Kohlenhydrat-defizientem Glykoprotein-Syndrom. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose Isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose-Isomerase-Mangel und Mannose-Therapie. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: Eine neue Manifestation des Kohlenhydratmangel-Glykoprotein-Syndroms, behandelbar mit Mannose. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG6 defective congenital disorder of glycosylation. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Airway mucus: From production to secretion. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Heparansulfat in der Skelettentwicklung, im Wachstum und in der Pathologie: der Fall der hereditären multiplen Exostosen. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Verschiedene nicht-entzündliche muskuloskelettale Erkrankungen. Hyperphosphatämische familiäre tumorale Calcinose (FGF23, GALNT3 und αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. Die Ablation des Galnt3-Gens führt zu niedrigen zirkulierenden Konzentrationen des intakten Fibroblasten-Wachstumsfaktors 23 (Fgf23) und Hyperphosphatämie trotz erhöhter Fgf23-Expression. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Eine Mutation in einem Gangliosid-Biosynthese-Enzym, ST3GAL5, führt zum Salz-& Pfeffer-Syndrom, einer neurokutanen Störung mit veränderter Glykolipid- und Glykoprotein-Glykosylierung. Hum Mol Genet 2014;23:418-33.
- Harlalka GV, Lehman A, Chioza B, et al. Mutationen in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. PIGA-Mutationen verursachen früh einsetzende epileptische Enzephalopathien und charakteristische Merkmale. Neurology. Neurology 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. Hypomorphe Promotormutation in PIGM verursacht vererbten Glycosylphosphatidylinositol-Mangel. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Hypomorphe Mutationen in PGAP2, das für ein GPI-Anker-Remodeling-Protein kodiert, verursachen autosomal-rezessive geistige Behinderung. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutationen in PIGO, einem Mitglied des GPI-Anker-Synthesewegs, verursachen Hyperphosphatasie mit mentaler Retardierung. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutationen im Glycosylphosphatidylinositol-Gen PIGL verursachen das CHIME-Syndrom. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J 1994;13:110-7.
- Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal rezessive dilatative Kardiomyopathie aufgrund von DOLK-Mutationen resultiert aus abnormaler Dystroglykan-O-Mannosylierung. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. Verbesserte diagnostische Ausbeute im Vergleich zu gezielten Gensequenzierungspanels legt eine Rolle für die Ganzgenomsequenzierung als Gentest der ersten Stufe nahe. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Currents in Contemporary Ethics. GINA, der ADA, und genetische Diskriminierung in der Beschäftigung. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Rezidivierende Thromboembolie bei einem Kind mit einer kongenitalen Glykosylierungsstörung (CDG) Typ Ib und Behandlung mit Mannose. Pediatr Hematol Oncol 2008;25:762-8.
- Mention K, Lacaille F, Valayannopoulos V, et al. Development of liver disease despite mannose treatment in two patients with CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Krampfanfälle und Stupor während intravenöser Mannose-Therapie bei einem Patienten mit CDG-Syndrom Typ 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Fucosesupplementierung bei Leukozytenadhäsionsmangel Typ II. Blood 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Inherited glycosylphosphatidyl inositol deficiency: a treatable CDG. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Oral D-Galactose supplementation in PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Galactose Supplementation in Patients with TMEM165-CDG Rescues the Glycosylation Defects. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8 deficiency: biochemical correction and major clinical improvement by manganese therapy. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. CDG Therapies: From Bench to Bedside. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Human glycosylation disorders. Marino PA, Editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev 2016;38:848-51.