- Visão geral
- Epidemiologia
- Classificação e nomenclatura bioquímica
- Genetics
- Patofisiologia
- Defeitos de glicosilação da proteína ligada ao N
- Defeitos de glicosilação ligados ao O e defeitos combinados de glicosilação ligados ao N e ao O
- Glosilcoilação lipídica e defeitos de biossíntese de âncoras GPI
- Aprovas clínicas
- Defeitos de glicosilação da proteína N-linked
- PMM2-CDG (deficiência de CDG-Ia, PMM2)
- MPI-CDG (CDG-Ib, deficiência de manosafosfato isomerase)
- ALG6-CDG (deficiência de glucosiltransferase 1)
- Defeitos de glicosilação ligados ao O e defeitos de glicosilação ligados ao N e O combinados
- Glicosilcosilação lipídica e defeitos de biossíntese da âncora GPI
- Diagnóstico
- Confunções de glicosilação de proteínas ligadas a N
- Defeitos de glicosilação ligados ao O e defeitos de glicosilação ligados ao N e O combinados
- Glosilação lipídica e defeitos de biossíntese de âncora GPI
- Análise molecular
- Gestão
- Terapias e prognóstico direcionados
- Agradecimentos
- Pé nota
Visão geral
A glicosilação é o processo de adição de resíduos de açúcar a proteínas e lipídios em diferentes vias celulares. Os distúrbios congênitos da glicosilação (CDG) são um grupo geneticamente e clinicamente heterogêneo de mais de cem doenças causadas por defeitos em várias etapas ao longo das vias de síntese ou modificação da glicosilação. A maioria dessas doenças monogenéticas são autossômicas recessivas em herança, mas formas autossômicas dominantes e ligadas ao X também foram descritas.
CDG tipicamente presentes com manifestações multissistêmicas, mais comumente atraso no desenvolvimento, falha no desenvolvimento, hipotonia, anormalidades neurológicas, hepatopatia, e coagulopatia. Os indivíduos afetados também podem se apresentar com doenças oculares, cutâneas e cardíacas, bem como com dismorfismos faciais. Apesar de alterações neurológicas e atrasos cognitivos serem observados na maioria dos indivíduos afetados, há certos casos e até tipos que não têm manifestações neurológicas. Dada a ampla etiologia clínica e genética da CDG, o diagnóstico clínico baseia-se em alto índice de suspeita em doença multissistêmica.
Análise de transferrina deficiente em carboidratos séricos (CDT) é o teste de triagem de primeira linha em pacientes com suspeita de CDG, mas é limitado na detecção de defeitos de glicosilação N com deficiência de ácido siálico. O teste de linha seguinte inclui a análise de glicosilano ligado ao dolicol e testes genéticos. O diagnóstico precoce deste grupo de doenças em crescimento exponencial é importante, uma vez que alguns CDG são tratáveis. O tratamento para defeitos de glicosilação é principalmente de apoio, embora estejam disponíveis terapias específicas para MPI-CDG, SLC35C1-CDG, PIGM-CDG, e PGM1-CDG. Detalhes sobre esses tratamentos estão na seção “Terapias direcionadas e Prognóstico” abaixo. O foco desta revisão será nos tipos mais comuns de DCG com fenótipos ou tratamentos reconhecíveis, sendo o público-alvo os prestadores de cuidados primários.
Epidemiologia
A incidência e prevalência de todos os tipos de DCG no agregado não foi bem estabelecida, embora pacientes de quase todas as origens étnicas e ambos os sexos sejam igualmente afetados. A prevalência estimada nas populações europeias e afro-americanas é de 1/10.000 com base nas frequências portadoras de variantes patogénicas conhecidas em 53 genes (1-4). A prevalência dos CDG mais comumente diagnosticados, PMM2-CDG, varia de 1/20.000 nas populações holandesas e 1/77.000 na Estônia com base em relatos isolados (5,6). Até o momento, menos de 100 casos foram relatados para a maioria dos tipos de CDG.
Classificação e nomenclatura bioquímica
Broadly, CDG atualmente são classificados em quatro categorias -(I) glicosilação N-linked, (II) glicosilação O-linked, (III) glicosilação combinada N-linked e O-linked/múltiplos, e (IV) lipídio e glicosilfosfatidilinositol (GPI) defeito de biossíntese de âncora.
O defeito de glicosilação da proteína N-linked PMM2-CDG, anteriormente conhecido como CDG tipo Ia, foi o primeiro CDG a ser relatado por Jaeken em 1980 e permanece de longe o CDG mais comum até hoje (7). O PMM2-CDG foi inicialmente denominado “síndrome de glicoproteína carboidrato deficiente” devido a múltiplas anormalidades da glicoproteína sérica observadas por focalização isoelétrica da transferrina sérica em indivíduos afetados. Historicamente, os CDG foram classificados por padrões de análise de isoformas de transferrina – padrões do tipo I foram atribuídos ao conjunto de glicoscópio ligado ao dolicol e defeitos de transferência localizados ao citoplasma ou ER, e padrões do tipo II foram atribuídos a defeitos de processamento no aparelho de Golgi. A partir deste ponto do ramo, os CDG foram então nomeados alfabeticamente em ordem de descoberta.
Com o advento do diagnóstico molecular generalizado, a nomenclatura CDG foi atualizada em 2008 para especificar a etiologia molecular da doença, refletindo o crescimento exponencial de vias e distúrbios que não se enquadravam perfeitamente nas categorias dicotômicas previamente estabelecidas. Atualmente, a nomenclatura CDG é denotada pelo nome do gene afetado (nãoitalicizado, nomes de genes em www.genenames.org), seguido por -CDG (por exemplo, PMM2-CDG) (8).
Genetics
A grande maioria das doenças congênitas da glicosilação são herdadas de forma autossômica recessiva, com uma mutação herdada de cada progenitor assintomático (portador). Os testes moleculares, geralmente com métodos de sequenciamento de próxima geração, são necessários para estabelecer um diagnóstico genético. Os testes parentais para a variante conhecida podem confirmar a herança versus a ocorrência de novo. Para herança autossômica recessiva, o risco de recorrência para irmãos e cada gravidez de um indivíduo afetado é de 25% por ser afetado, 50% por ser portador assintomático e 25% por não ser afetado.
Um punhado de CDG tem herança autossômica dominante (N-linked: GANAB-CDG, PRKCSH-CDG; O-linked: EXT1/EXT2-CDG, POFUT1-CDG, POGLUT1-CDG). Menos ligados em X (ALG13-CDG, SSR4-CDG, PIGA-CDG, SLC35A2-CDG, ATP6AP1-CDG). A maioria das formas de CDG dominantes e algumas ligadas ao X são devidas a mutações de novo. Doenças e genes específicos estão descritos abaixo na seção “fisiopatologia”.
Dados de mutação para todos os genes publicados para CDG estão disponíveis no Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Informações sobre variantes genéticas específicas estão disponíveis no Leiden Open Variation Database com ferramentas de patogenicidade silícica integradas (http://www.lovd.nl/3.0/home). Sinopses clínicas para genes específicos podem ser encontradas no Online Mendelian Inheritance in Man (http://www.omim.org/) ou em um escopo mais limitado em GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/). Dado o pequeno número de pacientes afetados para a maioria dos subtipos CDG, a correlação genótipo-fenótipo é difícil de estabelecer.
Patofisiologia
Acima de 130 tipos de CDG foram relatados até o momento (9,10). Dada a presença onipresente de vias de glicosilação, as CDG são extremamente diversas em sua patogênese bioquímica. Numerosas proteínas e lipídios (ou seja, esfingolipídios e glicolipídios) sofrem glicosilação com monossacarídeos e/ou oligossacarídeos, chamados coletivamente de glucanos, em diferentes compartimentos celulares. As suas localizações subcelulares são diversas, mas a maioria dos defeitos ocorre dentro do aparelho ER ou Golgi. As características clínicas e a etiologia genética dos CDG mais comuns por via está resumida na Tabela S1.
Amongst proteins, as glucanas são descritas por sua ligação com a cadeia de polipeptídeos – os glucanos estão ligados ao grupo amida da asparagina (Asn) enquanto os O-glycans estão ligados ao grupo hidroxila da serina ou da treonina. A síntese de N-glicanos requer a construção em etapas de açúcares ligados a nucleotídeos no citosol, montagem no retículo endoplasmático e processamento no aparelho de Golgi. Em contraste, a síntese de O-glicanos requer montagem mas nenhum processamento, portanto defeitos de O-glicosilação ocorrem predominantemente no aparelho de Golgi.
Defeitos de glicosilação da proteína ligada ao N
N-glicosilação envolve a fixação covalente das estruturas de carboidratos no grupo amida da cadeia lateral dos resíduos de Asn dentro de um consenso Asn-X-Ser/Thr local de aceitação, translocação do polipéptido do substrato para o retículo endoplasmático para remodelação, e modificação adicional da cadeia de N-glicanos dentro do Golgi (11,12). Defeitos em qualquer lugar ao longo da síntese, montagem e via de processamento podem levar a doenças clínicas.
PMM2-CDG é causado por variantes patogênicas no gene da fosfomanomutase 2 (PMM2), levando à deficiência da enzima PMM2 que catalisa a conversão citosólica do manose-6-fosfato em manose-1-fosfato na segunda etapa da síntese da manose de guanosina difosfato (GDP). A maioria dos pacientes abriga mutações heterozigóticas patogénicas de falta de sentido (www.lovd.nl/PMM2). A variante patogênica recorrente mais comum p.Arg141His é encontrada em aproximadamente 40% dos indivíduos afetados de ascendência européia, e p.Phe119Leu também é freqüentemente encontrado no norte da Europa (1). Correlações genótipo-fenótipo foram relatadas para PMM2-CDG (3,13,14).
MPI-CDG é um distúrbio autossômico recessivo causado por variantes patogênicas no gene da manose isomerase do fosfato (MPI) levando a isomerase fosfomannose deficiente (MPI). A MPI normalmente catalisa o primeiro passo da síntese do PIB-manose (ou seja, a conversão da fructose-6-fosfato em manose-6-fosfato), mas a fructose-6-fosfato não se acumula intracelularmente uma vez que também pode ser metabolizada pela via glicolítica. Portanto, embora bioquimicamente semelhante ao PMM2-CDG, o MPI-CDG não causa um envolvimento neurológico e multissistêmico tão significativo. O CDT é também o teste de triagem de escolha para MPI-CDG, que mostra um padrão do tipo 1. O diagnóstico pode então ser confirmado molecularmente ou por fibroblasto/leucócitos MPI.
ALG6-CDG é uma doença recessiva causada por mutações no ALG6, levando à fixação anormal de três moléculas de glicose em intermediários de manose ligados ao dolicol e hipoglicose a jusante de glicoproteínas séricas (15).
Defeitos de glicosilação ligados ao O e defeitos combinados de glicosilação ligados ao N e ao O
A glicosilação do O compreende a adição gradual de cadeias de carboidratos a resíduos de serina, treonina e hidroxilisina de proteínas por glicosiltransferases no aparelho de Golgi (16). Diversos tipos de glicosil latas O-linked foram associados à doença humana, nomeada pelo primeiro açúcar ligado ao resíduo do aminoácido (17).
Glosilcoilação lipídica e defeitos de biossíntese de âncoras GPI
Ancoras GPI são glicolípidos que sofrem montagem seqüencial no retículo endoplasmático e modificações dentro do Golgi. Os defeitos de biossíntese de âncoras GPI devido a deficiências enzimáticas são denominados alfabeticamente por ordem de descoberta e não cronologicamente por etapa de montagem. Uma vez sintetizadas, as âncoras GPI residem nas membranas plasmáticas e ligam centenas de proteínas da superfície celular, realizando uma infinidade de funções celulares. A maioria destas doenças são autossômicas recessivas, com a notável exceção da deficiência de PIGA ligado ao X.
Aprovas clínicas
Dada a omnipresença de vias de glicosilação, praticamente qualquer sistema orgânico pode estar envolvido na CDG, embora a maioria dos casos envolva anormalidades neurológicas. Alguns CDG apresentam ictiose, incluindo MPDU1-CDG, DOLK-CDG, SRD5A3-CDG (Figura 1), e PIGL-CDG (18,19). Quase todos os CDG presentes com doença multissistêmica nos primeiros anos de vida, exceto alguns que afetam apenas um único sistema orgânico (i.e, retina em DHDDS-CDG; junção neuromuscular em ALG2-CDG, ALG14-CDG, CFPT1-CDG; cérebro em ST3GAL3-CDG, TUSC3-CDG; pele ou músculo esquelético em POGLUT1-CDG, POFUT1-CDG; cartilagem em EXT1/EXT2-CDG; fígado em TMEM199-CDG; glóbulos vermelhos em SEC23B-CDG). A idade de início e gravidade pode variar desde a idade adulta neonatal até à idade adulta quase assintomática, e qualquer permutação entre elas. A constelação de sintomas mais comumente relatada inclui atraso no desenvolvimento, falha no desenvolvimento, hipotonia, anormalidades neurológicas, hipoglicemia e anormalidades variáveis do fígado, olhos, pele, gastrointestinais, imunológicas, esqueléticas e de coagulação (19).
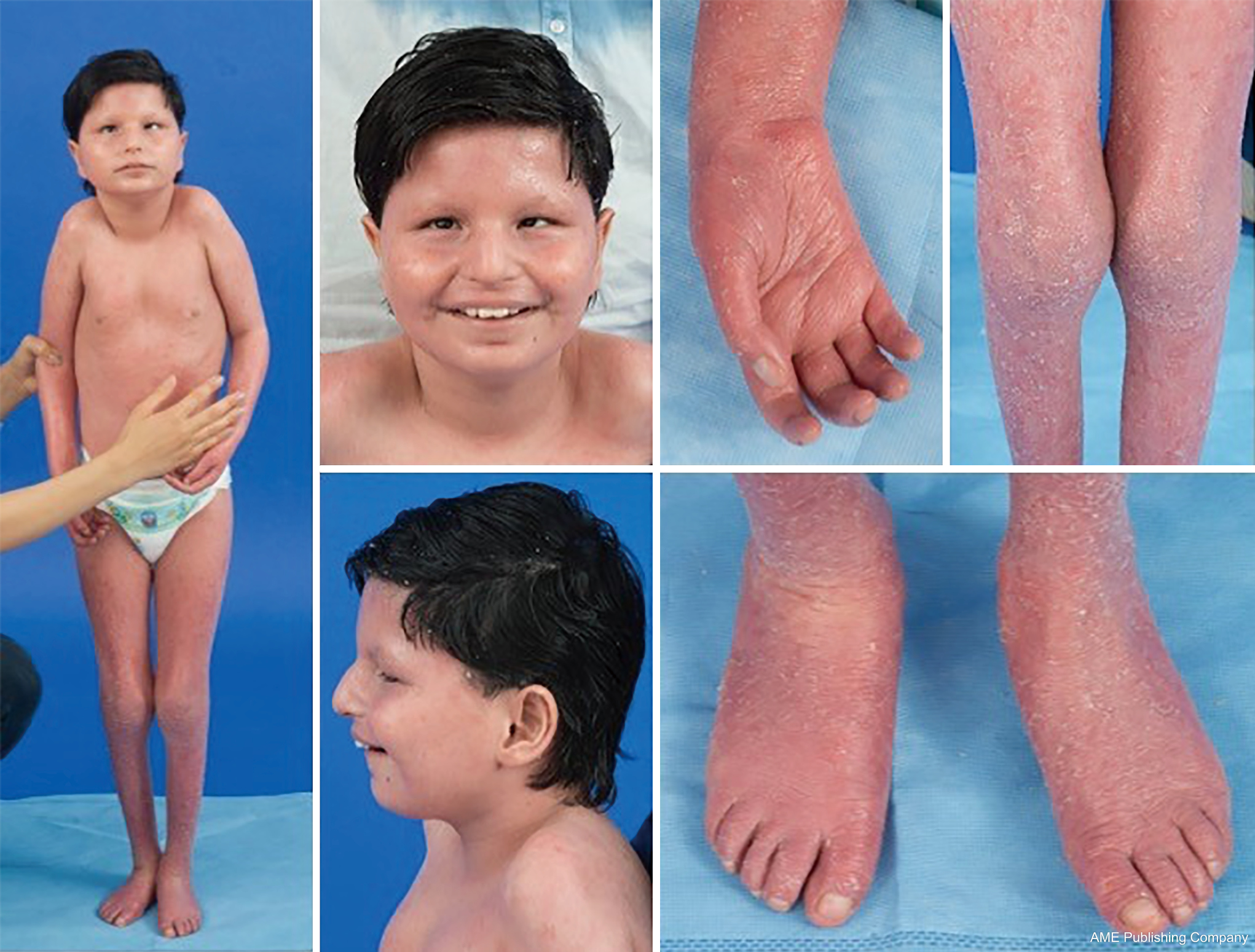
O fenótipo completo para muitos subtipos CDG ainda não foi totalmente delineado devido à raridade dos casos relatados. Portanto, a CDG deve ser considerada em qualquer cenário de doença multissistêmica, especialmente em casos com um componente neurológico ou atraso de desenvolvimento inespecífico com etiologia pouco clara.
Embora a fisiopatologia de sintomas múltiplos ainda não esteja elucidada, a relação entre certas vias de glicosilação e sintomas clínicos específicos tem sido esclarecida. Por exemplo, a falha em prosperar em muitos tipos de CDG é atribuível à hipoglicose e à formação deficiente de várias glicoproteínas dentro da via de crescimento da insulina, incluindo IGF-1, ALS e IGFBP-3 (20). Como entendemos melhor este grupo de distúrbios complexos, os GCD são cada vez mais reconhecidos em indivíduos com diagnósticos elusivos. Os envolvimentos do sistema orgânico de diferentes CDG estão resumidos na Tabela S1. Vamos discutir as características clínicas das formas e formas mais comuns com tratamentos direcionados para CDG abaixo.
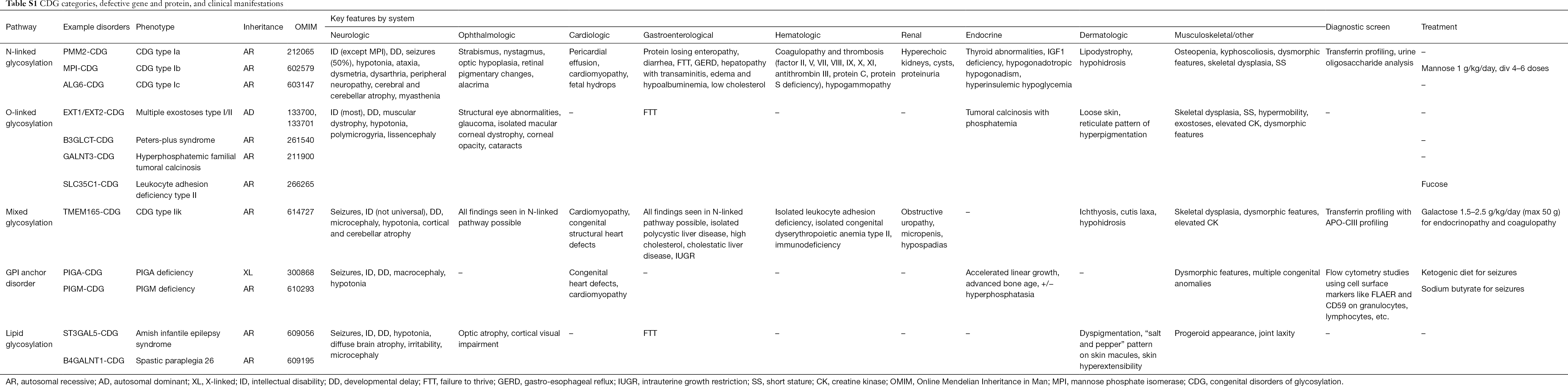
Tabela completa
Defeitos de glicosilação da proteína N-linked
Como a CDG mais comumente diagnosticada, o fenótipo dos distúrbios de glicosilação N-linked é frequentemente anunciado como a apresentação clássica. Entretanto, o espectro fenotípico do CDG é bastante diversificado, e muitos CDG podem não apresentar sintomas estereotipados associados ao PMM2-CDG.
PMM2-CDG (deficiência de CDG-Ia, PMM2)
PMM2-CDG é o CDG mais comum, com mais de 700 casos relatados em todo o mundo. Caracteriza-se por doença grave multissistêmica na infância, doença neurológica e atraso no desenvolvimento na infância, e/ou deficiência intelectual estável na idade adulta (21,22).
Na infância, PMM2-CDG apresenta anormalidades neurológicas tipicamente logo após o nascimento, a saber, estrabismo e movimentos oculares anormais, hipoplasia cerebelar, hipotonia, retardo psicomotor, ataxia, hipotonia e hiporreflexia. Os lactentes também podem apresentar doença hepática, síndrome nefrótica e cistos renais, derrame pericárdico e cardiomiopatia hipertrófica, insucesso no desenvolvimento e falência de múltiplos órgãos resultando em morte no primeiro ano de vida em até 20% dos indivíduos afetados (21,23-28).
Foi descrita uma constelação de características dismórficas em pacientes com PMM2-CDG (Figuras 2,3). Estes incluem um cerebelo hipoplásico, dismorfismos faciais (isto é, orelhas grandes e displásicas), mamilos invertidos e distribuição anormal do tecido adiposo sobre as nádegas ou região suprapúbica que pode se resolver com a idade (14,21,29-32). Os pacientes têm sido descritos como tendo um comportamento extrovertido e feliz. A apresentação é altamente variável, embora o estrabismo possa ser visto em mais de 70% dos pacientes afetados (21,23,33-35). Mamilos invertidos e almofadas de gordura anormais são vistos em cerca de 25-50% dos pacientes (36).

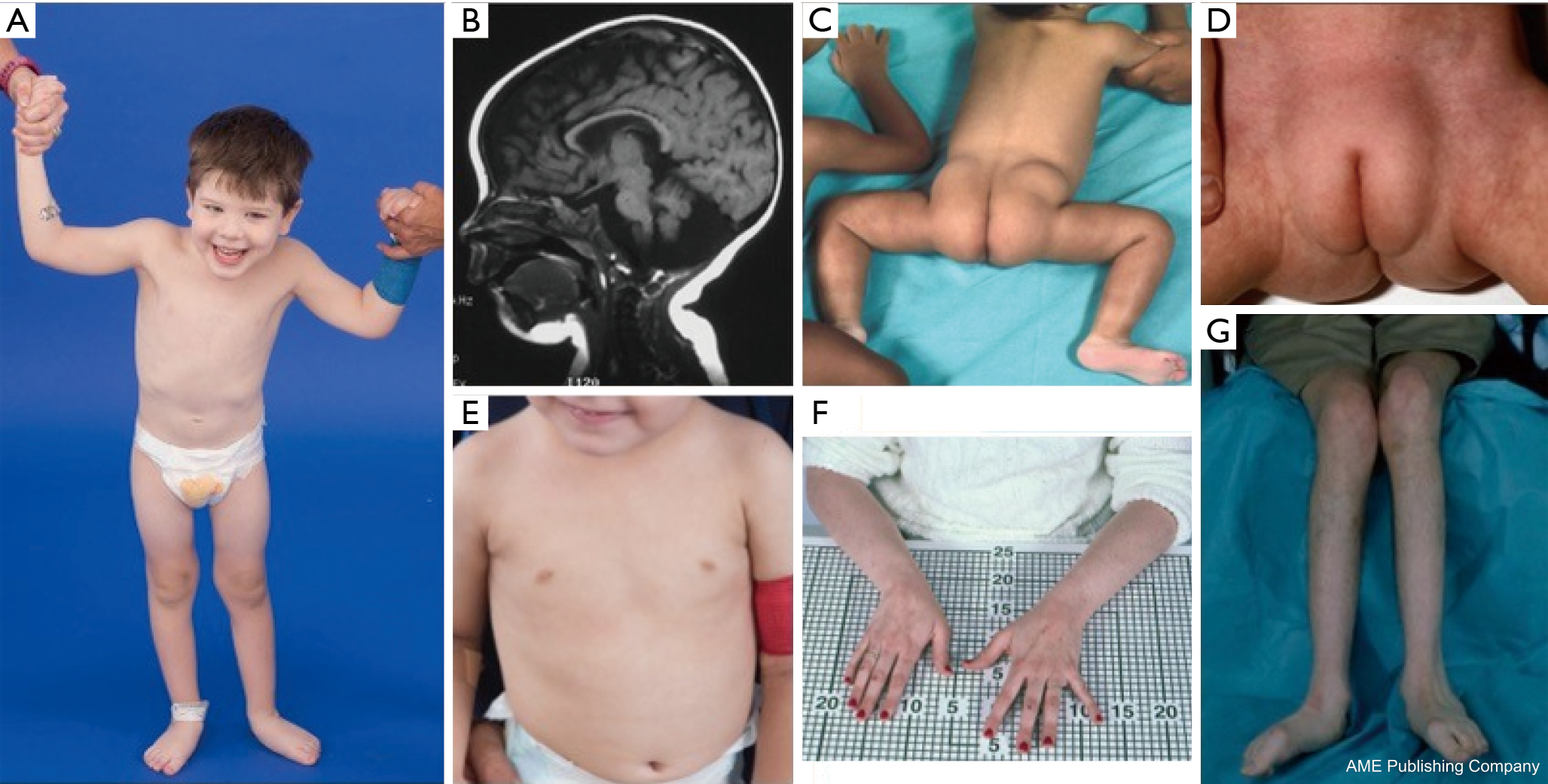
Na infância, os indivíduos afetados podem desenvolver retinite pigmentosa, episódios e convulsões semelhantes a acidentes vasculares cerebrais, atrasos na fala e motores, e neuropatia periférica. Constitucionalmente, os pacientes geralmente não conseguem prosperar devido a anormalidades alimentares e gastrointestinais e atraso no desenvolvimento global. Transaminases hepáticas elevadas sem consequência clínica podem ser observadas, as quais normalmente normalizam aos 5 anos de idade com flutuações ocasionais com doença (21,24). Biópsias hepáticas são raramente indicadas na CDG a menos que haja suspeita de fibrose hepática (1). Hipotireoidismo clínico é raro, mas pacientes com CDG devem ter seus hormônios tireoidianos e T4 livres medidos, que podem apresentar baixa ligação tireoidiana globulina (TBG) e elevações transitórias no hormônio estimulador da tireóide (TSH) (37). Malformações do fígado e dos canais biliares não foram relatadas em pacientes com PMM2-CDG.
Adultos com PMM2-CDG podem viver até as suas 7ª ou 8ª décadas com atraso cognitivo estável, neuropatia periférica e cifoscoliose torácica e espinhal progressiva com osteopenia ou osteoporose (34). A ataxia cerebelar é um sintoma cada vez mais reconhecido juntamente com o envolvimento multissistêmico (38-40). Anormalidades endócrinas incluindo hiperprolactinemia, liberação de hormônio de crescimento com hiperglicemia, resistência à insulina e hipoglicemia hiperinsulinêmica (41,42). Em mulheres afetadas, o hipogonadismo hipogonadotrópico pode levar à ausência de desenvolvimento sexual secundário ou ausência de ovários (41,43,44). Os pacientes podem estar em risco aumentado de trombose devido à diminuição dos fatores de coagulação sérica, incluindo fatores IV, IX e XI, antitrombina III, proteína C e proteína S (29).
MPI-CDG (CDG-Ib, deficiência de manosafosfato isomerase)
MPI-CDG é único porque os pacientes afetados têm pouco ou nenhum envolvimento neurológico e algumas manifestações da doença são tratáveis pela manose oral (2). Os sintomas são principalmente hepático-intestinais, sem características dismórficas ou atrasos cognitivos. Os pacientes normalmente apresentam vómitos recorrentes, hipoglicemia significativa, incapacidade de prosperar, enteropatia potencialmente fatal com perda de proteínas, alterações fibróticas hepáticas e dilatação do canal biliar (45-51). Os pacientes estão em risco aumentado de eventos trombóticos devido a baixas concentrações séricas de proteína C e S, e anti-trombina III.
ALG6-CDG (deficiência de glucosiltransferase 1)
ALG6-CDG é o segundo defeito mais comum de N-glicosilação caracterizado por fenótipo semelhante mas mais suave que o PMM2-CDG. Pacientes com ALG6-CDG têm falha no desenvolvimento, atraso no desenvolvimento, hipotonia, convulsões, estrabismo, ataxia, coagulopatia e dismorfismos faciais (ou seja, orelhas baixas, hipertelorismo e macroglossia). Semelhante ao MPI-CDG, eles também podem ter enteropatia por perda de proteínas. Além disso, os pacientes afetados podem ter anormalidades esqueléticas, incluindo braquidactilia e malformações e escoliose dos dedos. Os pacientes afetados normalmente não apresentam retinite pigmentosa ou hipoplasia cerebelar (52).
Defeitos de glicosilação ligados ao O e defeitos de glicosilação ligados ao N e O combinados
Devem à presença substancial de O-glycans em proteínas contendo mucina, incluindo glicosaminoglicanos (GAGs) e superfícies epiteliais (53), desordens de síntese de GAG tipicamente levam a displasias esqueléticas ou doença do tecido conjuntivo. Os pacientes afetados podem apresentar anormalidades musculoesqueléticas, cutâneas e articulares (por exemplo, frouxidão articular, exostoses múltiplas, condro/osteosarcomas), além de sintomas neurológicos (54-56). Por exemplo, a N-acetilgalactosaminiltransferase 3 (GALNT3) O-glycosylates o hormônio fosfatural, FGF23, prevenindo a clivagem proteolítica e permitindo a sua secreção intacta. A deficiência de GALNT3 leva à calcinose tumoral familiar, caracterizada por hiperfosfatemia e calcificações ectópicas (57,58).
Glicosilcosilação lipídica e defeitos de biossíntese da âncora GPI
Glycosphingolipids e seus derivados sialilados, gangliosides, são expressos principalmente por neurônios. Defeitos na quebra dos gangliosídeos levam ao acúmulo e às doenças bem caracterizadas do armazenamento lisossômico. No extremo oposto, defeitos na biossíntese dos gangliosídeos como ST3GAL5-CDG e B4GALNT1-CDG são extremamente raros e levam a doenças neurodegenerativas graves. Os pacientes podem apresentar paraplegia espástica, atraso intelectual grave, epilepsia e sintomas não neurológicos, incluindo displasia esquelética, características dismórficas e pigmentação anormal da pele (59,60).
Mutações em muitos genes dentro da via biossintética da âncora GPI causam uma variedade de anomalias congênitas múltiplas, incapacidade intelectual e epilepsia. O defeito de biossíntese GPI mais bem caracterizado, deficiência de PIGA ligado ao X, apresenta espasmos infantis com hipotonia, hipotonia, múltiplas anormalidades cerebrais e dismorfismos faciais. Os pacientes também podem ter doenças variáveis de pele, fígado, coração e renal (61-69). Algumas mutações dentro do PIGA causam a hemoglobinúria noturna paroxística (PNH), um distúrbio adquirido da falência da medula óssea (70,71).
Diagnóstico
Quando se suspeita clinicamente de um CDG, o primeiro passo é pedir testes bioquímicos de CDG no plasma ou soro, incluindo CDT e N-glycan. A análise sérica CDT e N-glicano só pode detectar defeitos de N-glicosilação, portanto não seriam úteis na diferenciação de defeitos isolados de O-glicosilação ou de âncora GPI. A análise isofórmica da transferrina foi originalmente obtida pelo foco isoelétrico da transferrina, já que a falha na síntese de N-glicanos causa deficiência parcial de ácido siálico, o que altera a carga sobre a transferrina sérica e posteriormente sua migração catódica sobre um campo eletroforético. Entretanto, a análise da transferrina e do N-glicano por espectrometria de massa substituiu em grande parte o foco isoelétrico pela identificação de alterações específicas nos oligossacarídeos por massa e carga (72).
Confunções de glicosilação de proteínas ligadas a N
Resultados da CDT transferrina de soro são relatados como a proporção de mono-oligosacarídeos/di-oligosacarídeos transferrinados, a-oligosacarídeo/di-oligosacarídeo transferrina, tri-sialo/di-oligosacarídeo transferrina, apolipoproteína CIII-1/apolipoproteína CIII-2, e apolipoproteína CIII-0/apolipoproteína CIII-2. Estes resultados quantitativos também virão com uma interpretação do padrão de achados.
A transferrina CDT tipo I é caracterizada pelo aumento das bandas di- e asialotransferrina, e indica defeitos na síntese de N-glicanos no citosol ou retículo endoplasmático. Um padrão tipo II é caracterizado pelo aumento das bandas de di- e asialotransferrina, e bandas tri- e/ou monoialotransferrina, e indica defeitos no processamento de N-glicanos no aparelho de Golgi (73).
Se for detectado um padrão CDT sérico de transferrina tipo I, a deficiência de PMM2 ou de MPI deve estar na vanguarda dos diferenciais, pois PMM2-CDG é o CDG mais comum e MPI-CDG é tratável e potencialmente fatal se não for tratado. Para diferenciar entre diagnósticos, deve ser feito um perfil de N-glycan, sequenciamento molecular ou testes enzimáticos. O diagnóstico de PMM2-CDG ou MPI-CDG é confirmado através de testes moleculares mostrando variantes patogênicas bialleicas em PMM2 ou MPI, seguidas pela atividade enzimática de PMM ou MPI em leucócitos ou fibroblastos se a patogenicidade das variantes genéticas for incerta. A análise N-glycan ou análise molecular diferenciaria a maioria dos ALG-CDG de PMM2 ou MPI-CDG (15).
Um padrão de CDT sérico de transferrina tipo II indica defeitos de Golgi, como a deficiência de N-acetilglucosaminiltransferase (GnT) II (CDG tipo IIA, MGAT2-CDG). A análise isoforma de Apolipoproteína CIII (Apo-CIII) é um teste complementar para um perfil CDT tipo II, pois mede defeitos de mucina tipo O-glicosilação no aparelho de Golgi. Há uma sensibilidade limitada para CDT ou Apo-CIII na detecção de CDG do tipo II. Assim, o perfil de N-glicano e O-glicano e o painel molecular ou sequenciamento de exomas devem ser realizados quando estes testes clínicos estiverem disponíveis. Os padrões de glicosilação por transferrina podem normalizar esporadicamente; portanto, testes repetidos podem ser indicados em pacientes com alto índice de suspeita. Os falsos positivos podem ser obtidos em pacientes com crise aguda de intolerância à frutose hereditária, galactosemia, doença hepática aguda e algumas infecções bacterianas. Nenhum dos testes bioquímicos CDG pode rastrear todos os CDG, portanto, mesmo na presença de resultados normais de rastreamento, podem ser realizados testes de painel genético molecular ou seqüenciamento de exomas para forte suspeita clínica. Por outro lado, a confirmação bioquímica e funcional dos achados genéticos moleculares também são essenciais, já que a maioria dos pacientes com CDG carrega pelo menos uma mutação leve e muitas vezes nova falha de mutação.
Defeitos de glicosilação ligados ao O e defeitos de glicosilação ligados ao N e O combinados
Diagnóstico depende do seqüenciamento molecular, já que a análise da isoforma de transferrina não detectaria defeitos isolados de glicosilação do O. Defeitos de glicosilação ligados ao N e O combinados podem ser detectados por CDT, análise ApoCIII, e análise de N-glicano e O-glicano plasmático.
Glosilação lipídica e defeitos de biossíntese de âncora GPI
Citometria de fluxo de granulócitos sanguíneos mede a expressão da superfície celular de proteínas ancoradas em GPI, como CD16 e CD24. A análise citométrica de fluxo de glóbulos brancos ou glóbulos vermelhos para certas proteínas de superfície celular com âncora GPI está disponível clinicamente como um teste para PNH devido a mutações adquiridas no gene PIGA. O teste PNH pode revelar anormalidades em outras deficiências de âncoras de GPI, mas o diagnóstico baseia-se principalmente na análise molecular.
Análise molecular
O maior rendimento diagnóstico para a CDG é o painel de sequenciamento genético baseado na próxima geração ou sequenciamento clínico de exomas (CES). O sequenciamento genético prova a sequência nucleotídica, ou ortografia “letra”, dos genes para determinar se há uma mudança que afeta a função do gene. O genoma humano consiste em 3 milhões de nucleotídeos, mas apenas 1-2% destes, chamados exônios, são traduzidos em um produto protéico funcional. O DNA não codificador restante intercalado entre os exons que não são traduzidos são chamados de introns (74). O CES examina quase todos os exons conhecidos dos aproximadamente 20.000 genes do genoma humano, o que representa uma minoria do material genético nos cromossomos, mas é mais provável que contenham variantes causadoras de doenças (patogênicas). Um resultado positivo significa que são identificadas variantes conhecidas causadoras de doença (ou seja, patogênicas), após as quais o diagnóstico, história natural, prognóstico, risco de recidiva e opções de tratamento podem ser discutidos. Um resultado negativo significa que não foram identificadas variantes patogénicas detectáveis. Variantes de significância desconhecida (também conhecidas como VUS) significam que embora tenham sido identificadas alterações genéticas, não há informação suficiente sobre a alteração genética específica para saber definitivamente se ela é causadora de doença. São esperadas variações no ADN de cada indivíduo, pelo que a realização de testes parentais simultâneos para comparação pode ajudar na interpretação laboratorial e clínica dos resultados. O rendimento diagnóstico do CES é estimado em 30-35% e está aumentando com o tempo à medida que a descoberta e o conhecimento sobre o genoma humano continuam a progredir (75-77). O CES está cada vez mais ordenado como o teste genético de primeira linha de escolha, dado o seu rápido tempo de resposta e o baixo custo relativo da quantidade de informação genética analisada. As limitações do CES incluem a falta de 100% de sensibilidade, a incapacidade de detectar certos tipos de alterações genéticas (por exemplo, deleções, duplicações, repetições de trinucleotídeos, mutações intrônicas profundas ou defeitos de metilação), e o fato de que um diagnóstico pode não fornecer informações adicionais sobre a doença ou o manejo da mudança.
No relato do CES, variantes patogênicas detectadas incidentalmente em genes associados a condições genéticas bem conhecidas podem ser relatadas como achados secundários (78). Esta lista de doenças recomendadas é curada pelo American College of Medical Genetics (ACMG). A Lei de Não Discriminação da Informação Genética (GINA) é uma consideração importante quando se decide se se deve ou não optar por participar ou não da aprendizagem de descobertas incidentais (79). A GINA protege os indivíduos do uso indevido de informação genética em seguros de saúde e emprego, mas não em seguros de vida. A GINA protege a seguinte informação genética: histórico médico familiar, testes de portadores, testes genéticos pré-natais, testes de susceptibilidade e preditivos e análise de tumores ou outras avaliações de genes, mutações ou alterações cromossômicas.
Gestão
A gestão da CDG depende em grande parte dos sintomas específicos do indivíduo. Sintomas recorrentes em pacientes com CDG incluem falha no desenvolvimento, atraso no desenvolvimento global, vômitos, episódios semelhantes a acidentes vasculares cerebrais e anormalidades esqueléticas. Coagulopatia clínica ou subclínica, endocrinopatia, hepatopatia e defeitos cardíacos também são comumente observados. Testes laboratoriais de base para estabelecer a extensão da doença e monitorização de rotina são recomendados, especialmente para PMM2-CDG. Estes incluem testes de função hepática, albumina sérica, testes de função tireoidiana incluindo T4 livre, proteína C, proteína S, antitrombina III, fator IX, urinálise e gonadotropinas séricas e hormônio de crescimento.
As imagens recomendadas incluem ecocardiograma, ultra-som renal, idade óssea, exame oftalmológico para avaliação do cristalino, retina, mobilidade ocular e pressão intra-ocular. Salvo indicação em contrário, as vacinas de rotina são recomendadas para adultos e crianças afetadas com CDG. Os títulos de anticorpos devem ser obtidos após a vacinação, pois os pacientes podem ter uma resposta imunogênica subótima. A repleção profilática dos fatores de coagulação antes de qualquer procedimento cirúrgico pode ser necessária se existirem deficiências na linha de base.
Avaliação genética clínica deve ser realizada para discutir os aspectos hereditários da DCG, bem como estabelecer um lar médico para esses pacientes complexos. O lar médico é tipicamente o serviço de genética bioquímica, embora os departamentos de genética, neurodesenvolvimento ou neurologia também tenham servido nesta capacidade se um serviço dedicado de genética bioquímica não estiver disponível. O encaminhamento de especialistas em gastroenterologia, hematologia, endocrinologia, suporte nutricional, fonoaudiologia, terapia ocupacional, física e de alimentação, ortopedia e medicina de reabilitação são freqüentemente necessários.
Terapias e prognóstico direcionados
Tratamento para a maioria dos tipos de CDG é amplamente favorável, com algumas exceções. O MPI-CDG é o tratamento mais eficaz de todos os tipos de CDG. A manose oral é convertida em manose-6-fosfato por hexokinases intracelulares, contornando assim o bloco enzimático e produzindo o substrato deficiente. A suplementação com manose começa tipicamente a 1 g/kg de peso corporal por dia, dividido em 4-6 doses por dia. Enquanto que a enteropatia potencialmente fatal com perda de proteína é especialmente responsiva ao tratamento da manose, a doença hepática no MPI-CDG pode continuar a progredir. Os sintomas clínicos melhoram rapidamente e a CDT transferrina normaliza-se ao longo dos meses, embora a doença hepática possa continuar a progredir com o tratamento (45,80,81).
Cautela deve ser exercida na suplementação da manose durante a gravidez, uma vez que a administração de manose em modelos de camundongos hipomórficos isomerase fosfomannose grávidas resultou em letalidade embrionária e cegueira em seus filhotes (82). Além disso, a manose intravenosa tem sido associada com diminuição da consciência e convulsões, que se resolveram com a administração de glicose (83).
O tratamento para PMM2-CDG é largamente favorável e baseado na sintomatologia. Entretanto, os próximos ensaios clínicos sobre a terapia de reposição do substrato de manose-1-fosfato estão atualmente em desenvolvimento.
Para outros CDG, vários açúcares simples orais têm sido investigados com o objetivo de melhorar teoricamente a hipoglicose. A fucose foi experimentada para SLC35C1-CDG e a galactose para PGM1-CDG e SLC35A2-CDG com resultados mistos (84). Foi demonstrado que a galactose D a 1,0-2,5 g/kg/dia (máx 50 gramas) melhora a hipoglicemia, coagulopatia e endocrinopatia no PGM1-CDG (85,86). Também foi demonstrado que a galactose melhora a endocrinopatia e a coagulopatia no TMEM165-CDG (87) e no SLC39A8-CDG. Uma melhora clínica considerável também foi relatada em pacientes com SLC39A8-CDG em 15-20 mg/kg/dia de MnSO4 (88). Ensaios clínicos estão em andamento para investigar a utilidade da N-acetilmannosamina (ManNAc) no GNE-CDG (89), e vários ensaios pré-clínicos estão em andamento para outros CDG (90).
Embora haja avanços médicos, existe mortalidade significativa para crianças com CDG no primeiro ano de vida por falência de múltiplos órgãos ou infecção grave (91). Lactentes com CDG podem apresentar doença fulminante multiorganismos, convulsões intratáveis ou hipoalbuminemia grave progredindo para anasarca. Alguns pacientes respondem a diurese agressiva e reposição de albumina enquanto outros são refractários ao tratamento. Foi demonstrado que o butirato de sódio melhora o controle das convulsões em CAD-CDG e PIGM-CDG (92). Também foi demonstrado que a dieta Ketogenética diminui a frequência de convulsões em alguns casos de PIGA-CDG (93). Durante episódios de AVC, a hidratação intravenosa e a manutenção da glicemia normal podem ser úteis enquanto a etiologia trombótica ou hemorrágica vascular subjacente é descartada.
Com o advento das técnicas de edição de genomas e uma melhor compreensão do mecanismo das doenças englobadas pelo guarda-chuva diagnóstico do CDG, o futuro do desenvolvimento terapêutico orientado permanece promissor.
Agradecimentos
Gostaríamos de agradecer a Lynne Wolfe, ARNP e Donna Krasnewich, MD, PhD por nos fornecerem fotos clínicas obtidas como parte das Investigações Clínicas e Básicas de Distúrbios Congênitos Conhecidos e Suspeitos de Glicosilação (NCT02089789). Gostaríamos também de agradecer a Jenny Thies, MS, LGC pela sua experiência em aconselhamento genético.
Funding: IJ Chang é apoiado pelos Institutos Nacionais de Saúde T32GM007454.
Pé nota
Conflitos de Interesse: Os autores não têm conflitos de interesse a declarar.
Consentimento Informado: O consentimento livre e esclarecido por escrito foi obtido dos pacientes para publicação deste manuscrito e de quaisquer imagens anexas.
- Jaeken J, Matthijs G. Desordens congênitas de glicosilação. Annu Rev Genomics Hum Genet 2001;2:129-51.
- Jaeken J, Matthijs G. Doenças congênitas da glicosilação: uma família de doenças em rápida expansão. Annu Rev Genomics Hum Genet 2007;8:261-78.
- Matthijs G, Schollen E, Bjursell C, et al. Mutações em PMM2 que causam doenças congênitas de glicosilação, tipo Ia (CDG-Ia). Hum Mutat 2000;16:386-94.
- Haeuptle MA, Hennet T. Desordens congênitas da glicosilação: uma atualização sobre defeitos que afetam a biossíntese de oligossacarídeos ligados ao dolicol. Hum Mutat 2009;30:1628-41.
- Vals MA, Pajusalu S, Kals M, et al. The Prevalence of PMM2-CDG in Estonia Based on Population Carrier Frequencies and Diagnosed Patients. JIMD Rep 2018;39:13-7.
- Schollen E, Kjaergaard S, Legius E, et al. Falta de equilíbrio de Hardy-Weinberg para a mutação de PMM2 mais prevalente na CDG-Ia (desordens congênitas de glicosilação tipo Ia). European Journal of Human Genetics 2000;8:367-71.
- Jaeken J, van Eijk HG, van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a new recognized genetic syndrome. Clin Chim Acta 1984;144:245-7.
- Jaeken J, Hennet T, Matthijs G, et al. Nomenclatura CDG: hora de uma mudança! Biochim Biophys Acta 2009;1792:825-6.
- Freeze HH, Chong JX, Bamshad MJ, et al. Solucionando distúrbios de glicosilação: abordagens fundamentais revelam caminhos complicados. Am J Hum Genet 2014;94:161-75.
- Jaeken J, Péanne R. O que há de novo na CDG? J Inherit Metab Dis 2017;40:569-86.
- Cylwik B, Naklicki M, Chrostek L, et al. Desordens congénitas de glicosilação. Parte I. Defeitos da proteína N-glicosilação. Acta Biochim Pol 2013;60:151-61.
- Helenius A, Aebi M. Funções intracelulares das latas de glicosilato N-linked. Science 2001;291:2364-9.
- Schiff M, Roda C, Monin ML, et al. Achados clínicos, laboratoriais e moleculares e dados de acompanhamento a longo prazo em 96 pacientes franceses com PMM2-CDG (desordem fosfomanomutase 2-congenital da glicosilação) e revisão da literatura. J Med Genet 2017;54:843-51.
- Barone R, Carrozzi M, Parini R, et al. Uma pesquisa nacional de PMM2-CDG na Itália: alta freqüência de uma variante neurológica leve associada à mutação L32R. J Neurol 2015;262:154-64.
- Dercksen M, Crutchley AC, Honey EM, et al. ALG6-CDG na África do Sul: Genótipo-Fenótipo Descrição de Cinco Pacientes Novel. JIMD Rep 2013;8:17-23.
- El-Battari A, Prorok M, Angata K, et al. Diferentes glicosiltransferases são diferentemente processadas para secreção, dimerização, e autoglicosilação. Glicobiologia 2003;13:941-53.
- Duncker IM, Asteggiano C, Freeze H. Transtornos congênitos da glicosilação. In: Mora-Montes H. Editor. Glycans: Bioquímica, Caracterização e Aplicações Desordens Congênitas de Glicosilação. Hauppauge: Nova Science, 2012;59-82.
- Jaeken J, Rymen D, Matthijs G. Desordens congênitas da glicosilação: outras causas de ictiose. Eur J Hum Genet 2014;22:444-4.
- Rymen D, Jaeken J. Manifestações cutâneas em CDG. J Inherit Metab Dis 2014;37:699-708.
- Miller BS, Khosravi MJ, Patterson MC, et al. Sistema IGF em crianças com doenças congénitas de glicosilação. Clin Endocrinol (Oxf) 2009;70:892-7.
- de Lonlay P, Seta N, Barrot S, et al. Um amplo espectro de apresentações clínicas em doenças congênitas de glicosilação I: uma série de 26 casos. J Med Genet 2001;38:14-9.
- Kjaergaard S, Müller J, Skovby F. Crescimento pré-púbere em distúrbios congênitos da glicosilação do tipo Ia (CDG-Ia). Arch Dis Child 2002;87:324-7.
- Grünewald S, Imbach T, Huijben K, et al. Características clínicas e bioquímicas da desordem congênita da glicosilação tipo Ic, o primeiro defeito reticulum endoplasmático reconhecido na síntese de N-glicanos. Ann Neurol 2000;47:776-81.
- Marquardt T, Denecke J. Desordens congênitas de glicosilação: revisão de suas bases moleculares, apresentações clínicas e terapias específicas. Eur J Pediatr 2003;162:359-79.
- Kristiansson B, Stibler H, Hagberg B, et al. CDGS-1–uma doença metabólica hereditária recentemente descoberta. Manifestações de múltiplos órgãos, incidência 1/80.000, difícil de tratar. Lakartidningen 1998;95:5742-8.
- Marquardt T, Hülskamp G, Gehrmann J, et al. Grave isquemia miocárdica transitória causada por cardiomiopatia hipertrófica em um paciente com distúrbio congênito de glicosilação tipo Ia. Eur J Pediatr 2002;161:524-7.
- Romano S, Bajolle F, Valayannopoulos V, et al. Defeitos cardíacos conotruncais em três pacientes com distúrbio congênito da glicosilação do tipo Ia (CDG Ia). J Med Genet 2009;46:287-8.
- Truin G, Guillard M, Lefeber DJ, et al. Acumulação de líquido pericárdico e abdominal em distúrbio congênito de glicosilação tipo Ia. Mol Genet Metab 2008;94:481-4.
- Krasnewich D, O’Brien K, Sparks S. Características clínicas em adultos com doenças congênitas de glicosilação do tipo Ia (CDG-Ia). Am J Med Genet 2007;145C:302-6.
- Krasnewich D, Gahl WA. Síndrome da glicoproteína carboidrato deficiente. Adv Pediatr 1997;44:109-40.
- Enns GM, Steiner RD, Buist N, et al. Características clínicas e moleculares da desordem congênita da glicosilação em pacientes com padrão de sialotransferrina tipo 1 e diversas origens étnicas. J Pediatr 2002;141:695-700.
- Pérez J de J, Udeshi ND, Shabanowitz J, et al. A modificação O-GlcNAc da proteína do revestimento do vírus Potyvirus Plum pox virus aumenta a infecção viral. Virologia 2013;442:122-31.
- Erlandson A, Bjursell C, Stibler H, et al. Pacientes escandinavos CDG-Ia: correlação genótipo/fenótipo e origem geográfica das mutações fundadoras. Hum Genet 2001;108:359-67.
- Monin ML, Mignot C, De Lonlay P, et al. 29 pacientes adultos franceses com distúrbio congênito de glicosilação PMM2: resultado do fenótipo pediátrico clássico e representação de um fenótipo tardio. Orphanet J Rare Dis 2014;9:207.
- Thompson DA, Lyons RJ, Russell-Eggitt I, et al. Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG. J Inherit Metab Dis 2013;36:1039-47.
- Kjaergaard S, Schwartz M, Skovby F. Desordem congénita da glicosilação tipo Ia (CDG-Ia): espectro fenotípico do genótipo R141H/F119L. Arch Dis Child 2001;85:236-9.
- Mohamed M, Theodore M, Claahsen-van der Grinten H, et al. Função tireoidiana em PMM2-CDG: abordagem diagnóstica e manejo proposto. Mol Genet Metab 2012;105:681-3.
- Schoffer KL, O’Sullivan JD, McGill J. Desordem congénita de glicosilação tipo Ia que se apresenta como ataxia cerebelar precoce num adulto. Mov Disord 2006;21:869-72.
- Barone R, Sturiale L, Fiumara A, et al. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007;30:107-7.
- Coman D, McGill J, MacDonald R, et al. Desordem congênita de glicosilação tipo 1a: três irmãos com um fenótipo neurológico leve. J Clin Neurosci 2007;14:668-72.
- Miller BS, Freeze HH. Novas desordens no metabolismo dos carboidratos: desordens congênitas da glicosilação e seu impacto no sistema endócrino. Rev Endocr Metab Disord 2003;4:103-13.
- Shanti B, Silink M, Bhattacharya K, et al. Desordem congênita da glicosilação tipo Ia: heterogeneidade na apresentação clínica desde falha multivisceral até hipoglicemia hiperinsulinémica como sintomas principais em três lactentes com deficiência de fosfomannomutase. J Inherit Metab Dis 2009;32 Suppl 1:S241-51.
- de Zegher F, Jaeken J. Endocrinologia da síndrome da glicoproteína carboidrato deficiente tipo 1 desde o nascimento até a adolescência. Pediatr Res 1995;37:395-401.
- Kristiansson B, Stibler H, Wide L. Função Gonadal e hormônios glicoproteicos na síndrome da glicoproteína carboidrato deficiente (CDG). Acta Paediatr 1995;84:655-9.
- de Lonlay P, Seta N. O espectro clínico da deficiência de isomerase fosfomannose, com uma avaliação do tratamento da manose para a CDG-Ib. Biochim Biophys Acta 2009;1792:841-3.
- Marques-da-Silva D, Francisco R, Webster D, et al. Complicações cardíacas de distúrbios congênitos da glicosilação (CDG): uma revisão sistemática da literatura. J Inherit Metab Dis 2017;40:657-72.
- de Koning TJ, Toet M, Dorland L, et al. Hidrops fetalis não imunes recorrentes associados à síndrome da glicoproteína carboidrato deficiente. J Inherit Metab Dis 1998;21:681-2.
- Jaeken J, Matthijs G, Saudubray JM, et al. Phosphomannose isomerase deficiente: uma síndrome de glicoproteína carboidrato deficiente com apresentação hepático-intestinal. Am J Hum Genet 1998;62:1535-9.
- Niehues R, Hasilik M, Alton G, et al. Síndrome de glicoproteína carboidrato deficiente tipo Ib. Deficiência de fosfomannose isomerase e terapia de manose. J Clin Invest 1998;101:1414-20.
- Babovic-Vuksanovic D, Patterson MC, Schwenk WF, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr 1999;135:775-81.
- de Lonlay P, Cuer M, Vuillaumier-Barrot S, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: Uma nova manifestação de síndrome de glicoproteína carboidrato deficiente, tratável com manose. J Pediatr 1999;135:379-83.
- Jaeken J, Lefeber D, Matthijs G. Cartão genético de utilidade clínica para: ALG6 desordem congénita defeituosa de glicosilação. Eur J Hum Genet 2015;23:17.
- Williams OW, Sharafkhaneh A, Kim V, et al. Muco das vias aéreas: Da produção à secreção. Am J Respir Cell Mol Biol 2006;34:527-36.
- Rafaelsen S, Johansson S, Ræder H, et al. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperphostasis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet 2014;15:98.
- Huegel J, Sgariglia F, Enomoto-Iwamoto M, et al. Sulfato de Heparan no desenvolvimento, crescimento e patologia do esqueleto: o caso de exostoses múltiplas hereditárias. Dev Dyn 2013;242:1021-32.
- Cartault F, Munier P, Jacquemont ML, et al. Expandindo o espectro clínico da deficiência de B4GALT7: mutação homozigotos p.R270C com efeito fundador causa síndrome de Larsen da Ilha da Reunião. Eur J Hum Genet 2015;23:49-53.
- Farrow EG, Imel EA, White KE. Miscelânea de condições musculoesqueléticas não-inflamatórias. Calcinose tumoral familiar hiperfosfatêmica (FGF23, GALNT3 e αKlotho). Best Pract Res Clin Rheumatol 2011;25:735-47.
- Ichikawa S, Sorenson AH, Austin AM, et al. A ablação do gene Galnt3 leva a baixas concentrações de fator de crescimento 23 (Fgf23) de fibroblastos intactos e hiperfosfatemia, apesar do aumento da expressão de Fgf23. Endocrinology 2009;150:2543-50.
- Boccuto L, Aoki K, Flanagan-Steet H, et al. Uma mutação em uma enzima biossintética ganglioside, ST3GAL5, resulta em sal & síndrome da pimenta, uma desordem neurocutânea com glicolipídeo alterado e glicosilação da glicoproteína. Hum Mol Genet 2014;23:418-33.
- Harlalalka GV, Lehman A, Chioza B, et al. As mutações em B4GALNT1 (GM2 synthase) estão subjacentes a uma nova desordem de biossíntese de ganglioside. Cérebro 2013;136:3618-24.
- Kato M, Saitsu H, Murakami Y, et al. Mutações em PIGA causam encefalopatias epilépticas precoces e características distintivas. Neurologia. Neurologia 2014;82:1587-96.
- Maydan G, Noyman I, Har-Zahav A, et al. Múltiplas anomalias congênitas – síndrome de hipotonia convulsiva é causada por uma mutação em PIGN. J Med Genet 2011;48:383-9.
- Almeida AM, Murakami Y, Layton DM, et al. A mutação promotora hipomórfica em PIGM causa deficiência de glicosilfosfatidilinositol hereditário. Nat Med 2006;12:846-51.
- Hansen L, Tawamie H, Murakami Y, et al. Mutações hipomórficas no PGAP2, codificando uma proteína de remodelação do GPI, causam deficiência intelectual autossômica recessiva. Am J Hum Genet 2013;92:575-83.
- Johnston JJ, Gropman AL, Sapp JC, et al. O fenótipo de uma mutação na linha germinal do PIGA: o gene mutadoomaticamente na hemoglobinúria nocturna paroxística. Am J Hum Genet 2012;90:295-300.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutações em PIGO, um membro da via de síntese GPI-anchor-síntese, causam hiperfosfatástase com retardo mental. Am J Hum Genet 2012;91:146-51.
- Kvarnung M, Nilsson D, Lindstrand A, et al. Uma nova síndrome de deficiência intelectual causada por deficiência de âncora GPI devido a mutações homozigotos em PIGT. J Med Genet 2013;50:521-8.
- Ng BG, Hackmann K, Jones MA, et al. Mutações no gene glicosilfosfatidilinositol PIGL causam a síndrome de CHIME. Am J Hum Genet 2012;90:685-8.
- Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S, et al. O espectro genotípico e fenotípico da deficiência de PIGA. Orphanet J Rare Dis 2015;10:23.
- Bessler M, Mason PJ, Hillmen P, et al. A hemoglobinúria nocturna paroxística (PNH) é causada por mutações somáticas no gene PIGA. EMBO J 1994;13:110-7.
- Brodsky RA. Hemoglobinúria paroxística noturna. Sangue 2014;124:2804-11.
- Sturiale L, Barone R, Garozzo D. O impacto da espectrometria de massa no diagnóstico de distúrbios congênitos da glicosilação. J Inherit Metab Dis 2011;34:891-9.
- Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet 2011;7:e1002427.
- Sakharkar MK, Chow VTK, Kangueane P. Distribuições de exons e introns no genoma humano. Em Silico Biol (Gedrukt) 2004;4:387-93.
- Yang Y, Muzny DM, Reid JG, et al. Sequenciação clínica de exônomos inteiros para o diagnóstico de distúrbios mendelianos. N Engl J Med 2013;369:1502-11.
- Lionel AC, Costain G, Monfared N, et al. A melhoria do rendimento diagnóstico em comparação com os painéis de sequenciação de genes alvo sugere um papel para a sequenciação do genoma inteiro como um teste genético de primeiro nível. Genet Med 2018;20:435-43.
- Dragojlovic N, Elliott AM, Adam S, et al. O custo e rendimento diagnóstico do sequenciamento de exomas para crianças com suspeita de distúrbios genéticos: um estudo de benchmarking. Genet Med 2018;20:1013-21.
- Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249-55.
- Rothstein MA. Correntes na ética contemporânea. GINA, a ADA, e a discriminação genética no emprego. J Law Med Ethics 2008;36:837-40.
- Tamminga RY, Lefeber DJ, Kamps WA, et al. Tromboembolismo recorrente em uma criança com uma doença congênita de glicosilação (CDG) tipo Ib e tratamento com manose. Pediatr Hematol Oncol 2008;25:762-8.
- Menção K, Lacaille F, Valayannopoulos V, et al. Desenvolvimento de doença hepática apesar do tratamento com manose em dois pacientes com CDG-Ib. Mol Genet Metab 2008;93:40-3.
- Sharma V, Nayak J, DeRossi C, et al. Os suplementos de manose induzem letalidade embrionária e cegueira em ratos hipomórficos fosfomannose isomerase. FASEB J 2014;28:1854-69.
- Schroeder AS, Kappler M, Bonfert M, et al. Convulsões e estupor durante terapia de manose intravenosa em um paciente com síndrome CDG tipo 1b (MPI-CDG). J Inherit Metab Dis 2010;33 Suppl 3:S497-502.
- Etzioni A, Tonetti M. Suplemento de Fucose em deficiência de adesão leucocitária tipo II. Sangue 2000;95:3641-3.
- Almeida A, Layton M, Karadimitris A. Deficiência hereditária de inositol glicosilfosfatidil: um CDG tratável. Biochim Biophys Acta 2009;1792:874-80.
- Wong SY, Gadomski T, van Scherpenzeel M, et al. Suplementação oral de D-galactose em PGM1-CDG. Genet Med 2017;19:1226-35.
- Morelle W, Potelle S, Witters P, et al. Suplementação com Galactose em Pacientes com TMEM165-CDG Resgata os Defeitos de Glicosilação. J Clin Endocrinol Metab 2017;102:1375-86.
- Park JH, Hogrebe M, Fobker M, et al. SLC39A8 deficiência: correção bioquímica e melhora clínica importante pela terapia com manganês. Genet Med 2018;20:259-68.
- Niethamer TK, Yardeni T, Leoyklang P, et al. Terapias orais de monossacarídeos para reverter a hiposialilação renal e muscular em um modelo de miopatia GNE de camundongo. Mol Genet Metab 2012;107:748-55.
- Brasil S, Pascoal C, Francisco R, et al. Terapias CDG: Da Bancada à Cabeceira. Int J Mol Sci 2018;19:1304.
- Krasnewich D. Distúrbios de Glicosilação Humana. Marino PA, editor. Cancer Biomark 2014;14:3-16.
- Almeida AM, Murakami Y, Baker A, et al. Terapia orientada para a deficiência de GPI hereditária. N Engl J Med 2007;356:1641-7.
- Joshi C, Kolbe DL, Mansilla MA, et al. Ketogenic diet – A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev 2016;38:848-51.