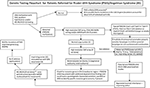
Graphical Abstract. Organigrama testelor genetice pentru pacienții trimiși pentru sindromul Prader-Willi (PWS)/ sindromul Angelman (AS). *Excludeți translocațiile sau inversiunile Chr15 prin studii cromozomiale de rutină; luați în considerare alte tulburări genetice legate de obezitate; poate fi necesară depistarea ADN-ului sindromului X fragil pentru expansiunea repetată a genei FMR1 sau teste genetice avansate cu secvențiere de generație următoare (NGS) pentru FMR1 sau alte variante genetice candidate utilizând secvențierea întregului exom (WES) sau secvențierea întregului genom (WGS; de exemplu, cauze monogenice ale obezității). **Poate fi utilizată pentru a verifica starea de metilare a altor gene cu amprentă Chr15; ddPCR, droplet digital PCR poate fi utilizată pentru screeningul mozaicismului.
Introducere
Tulburările de amprentă a cromozomului 15 includ sindroamele Prader-Willi (PWS) și Angelman (AS) (1-6) și duplicațiile cromozomului 15q. Diagnosticul de PWS sau AS depinde de părintele de origine și dacă expresia este limitată în mod aberant la genele imprimate materne sau paterne. Duplicarea 15q este cauzată de o copie suplimentară a regiunii 15q11.2-q13 de origine maternă, care poate duce la convulsii, probleme cognitive și comportamentale, inclusiv tulburări de spectru autist (TSA), dar nu la un fenotip PWS sau AS. PWS apare ca urmare a pierderii genelor imprimate la nivel matern și exprimate la nivel patern din regiunea cromozomului 15q11-q13, în timp ce AS este cauzată de pierderea genelor imprimate și exprimate la nivel matern din această regiune, cu impact specific asupra genei UBE3A. Datorită naturii imprimate a genelor responsabile, atât erorile genetice, cât și cele epigenetice pot fi cauzale.
În 1989, s-a constatat că cei care prezentau atât sindromul PWS, cât și cei fără deleție, aveau disomie maternă 15 sau ambele 15 de la mamă atunci când s-au folosit markeri ADN polimorfici din regiunea proximală 15q11-q13 (7). Ulterior, la mijlocul anilor 1990, dezvoltarea sondelor de ADN prin hibridizare fluorescentă in situ (FISH) a fost utilizată pentru a identifica delețiile din regiunea 15q11-q13 atât în PWS, cât și în AS. Testele ADN de metilare au fost dezvoltate în această perioadă de timp și s-a observat un model de metilare anormal în PWS și AS. Testul ADN de metilare are o precizie de ~99% în identificarea diagnosticului de PWS, dar nu va identifica clasa moleculară individuală de PWS (2). Pentru AS, testul de metilare a ADN-ului identifică ~80% dintre indivizi, dar, din nou, nu face distincția între clasele moleculare sau nu detectează o mutație în gena UBE3A care cauzează AS.
Tehnologia microarray a fost dezvoltată la începutul și mijlocul anilor 2000 și a avansat randamentul diagnosticului. Acum, noile microarrays SNP includ peste două milioane de sonde ADN și sunt utile în detectarea subtipurilor de deleție și a subclaselor UPD15. Alte tehnologii, cum ar fi droplet digital PCR (ddPCR), cuantifică numărul de copii folosind sonde de ADN ale cromozomului 15 și pot diagnostica defectele genetice în PWS sau AS (8). Mai mult, microplăcile SNP pot identifica LOH-uri definite ca având o dimensiune >8 Mb și, atunci când sunt prezente pe cromozomul 15, susțin diagnosticul de disomie 15 maternă sau disomie 15 paternă în prezența unui model anormal de metilare a ADN-ului pentru PWS sau, respectiv, AS. Confirmarea defectului de amprentă poate necesita nu numai microplăci SNP pentru a identifica microdelețiile mici, ci și probe de ADN parental cu genotipare pentru a identifica prezența unei moșteniri normale (biparentale) a cromozomului 15s care să susțină prezența unui defect de amprentă cu epimutație în PWS sau AS, influențând astfel riscurile de recurență. Diferențierea unei microdeleții IC de un statut de epimutație fără deleție este importantă din punct de vedere clinic pentru familii, deoarece există un risc de recurență de 50% pentru copiii suplimentari în cazul în care se găsește o microdeleție IC la părinte (9).
Există peste o duzină de gene și transcripții în regiunea 15q11-q13 care par să joace un rol în cauzalitatea PWS și/sau AS. Genele și transcriptele incluse în zona de la punctul de ruptură proximal 15q11.2 BP1 și punctul de ruptură distal 15q13 BP3 sunt TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, ARN-uri non-codificatoare (SNORD), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 și HERC2. Genele imprimate MRKN3, MAGEL2, NDN, NIPAP1 și SNURF-SNRPN sunt exprimate paternal și, atunci când sunt perturbate, pot cauza caracteristici ale PWS . De exemplu, mutațiile genei MAGEL2 pot cauza hipotonie neonatală, întârziere în dezvoltare, artrogripoză, trăsături autiste, supt slab și obezitate . De asemenea, au fost raportați pacienți cu trăsături de PWS ca urmare a unor deleții mici ale transcriptului non-codificator SNORD116 (12) și a altor deleții similare din regiune (10, 13).
Ne-am concentrat pe AS și PWS în acest raport, deoarece ambele sindroame sunt detectate prin intermediul testelor de metilare a ADN-ului, care permit determinarea alelei active a genei parentale și diagnosticarea definitivă la persoanele cu PWS și la majoritatea persoanelor cu AS (2). Cu toate acestea, testul de metilare a ADN nu va identifica clasa moleculară în niciunul dintre cele două sindroame. Analiza cromozomială de înaltă rezoluție a fost dezvoltată și utilizată la începutul anilor 1980 și a devenit un test standard de laborator bazat pe genetică pentru a evalua deleția cromozomului 15q11-q13 identificată la majoritatea pacienților cu PWS la acea vreme (14) și, mai târziu, pentru AS. Originea paternă a deleției 15q11-q13 a fost raportată în 1983 (15) și s-a constatat că este de novo, dar dimensiunea deleției 15q11-q13 sau tipul (tipică vs. atipică) nu a putut fi determinată. Diagnosticul precis și precoce cu identificarea clasei moleculare este esențial nu numai pentru a confirma diagnosticul clinic, ci și pentru consilierea genetică, pentru a informa îngrijirea și tratamentul și pentru a ghida așteptările. În intenția studiilor clinice în curs de desfășurare, o mai bună înțelegere a etiologiei moleculare poate avea un impact asupra oportunităților de participare a pacienților. Mai mult, studiile iminente includ oligonucleotide antisens pentru reactivarea copiei paterne reduse la tăcere a cromozomului 15 la persoanele cu AS.
PWS și AS sunt tulburări complexe rare de neurodezvoltare datorate unor erori în imprimarea genomică. PWS este recunoscut ca fiind cea mai frecventă cauză genetică a obezității care pune viața în pericol, dacă este lăsată necontrolată (2, 4, 6). Există trei clase moleculare recunoscute de PWS, inclusiv o deleție paternă 15q11-q13 cu o dimensiune de aproximativ 5-6 Mb (60% din cazuri) și disomie maternă 15 (UPD15) în care ambii cromozomi 15 sunt moșteniți de la mamă (36%) care provine din trisomia 15 cu pierderea cromozomului 15 patern la începutul sarcinii, ceea ce duce la doi cromozomi 15 de la mamă (16). Cea de-a treia clasă este un defect al centrului de amprentă. Dacă pe alela paternă este prezentă o microdeleție sau o epimutație a centrului de amprentare (IC), care controlează starea de expresie a unor gene imprimate selectate pe cromozomul 15, atunci apare PWS. Acest defect de amprentare este întâlnit la 4% dintre persoanele cu PWS (8, 16). Majoritatea cazurilor de SPW sunt sporadice, cu o echitate aproximativă între grupurile etnice și sex. Prevalența estimată a SPW este de unul la 10.000 până la unul la 30.000 (2). Numărul indivizilor din întreaga lume cu PWS este considerat a fi de ~400.000, cu aproximativ 20.000 de indivizi care trăiesc în SUA (2, 17).
PWS se caracterizează prin hipotonie infantilă, un reflex de supt slab cu dificultăți de hrănire, statură scurtă cu mâini și picioare mici, hipogonadism secundar deficiențelor hormonale, dizabilitate intelectuală ușoară, probleme de comportament și hiperfagie, adesea cu debut între 6 și 8 ani, care persistă la vârsta adultă și care duce la obezitate dacă nu sunt instituite controale de mediu. În timpul copilăriei, se observă trăsături cranio-faciale caracteristice, inclusiv un diametru bifrontal îngust, strabism, nas mic întors în sus cu buza superioară subțire și colțuri ale gurii întoarse în jos, salivă lipicioasă și hipoplazie a smalțului (2, 4, 6, 18). Cogniția este, în general, redusă în funcție de mediul familial, iar problemele de comportament care încep în copilărie includ automutilarea (scobirea pielii), accese de furie, încăpățânare și accese de furie, problemele psihiatrice apărând în această perioadă sau mai târziu, în adolescență sau la vârsta adultă tânără (2). Problemele comportamentale includ anxietatea, tulburările de dispoziție, psihoza și autismul care se pot corela cu subtipuri genetice specifice PWS sau clase moleculare (19).
În mod istoric, PWS este împărțit în două stadii clinice, cu eșecul de dezvoltare în timpul copilăriei reprezentând primul stadiu clinic și hiperfagia cu debutul obezității reprezentând al doilea stadiu (2). Ulterior, au fost descrise faze nutriționale pentru această tulburare genetică legată de obezitate și includ: Faza 0 cu scăderea mișcărilor fetale și retard de creștere in utero, urmată de Faza 1 legată de hipotonie, eșecul de dezvoltare cu dificultăți de hrănire, Faza 2 care începe la vârsta de ~2 ani când se observă pentru prima dată creșterea în greutate și Faza 3 când lipsa de sațietate este însoțită de căutare de alimente și hiperfagie care duce la obezitate, dacă nu este controlată extern. Faza 3 începe în jurul vârstei de 6-8 ani (20).
Sindromul Angelman se caracterizează prin întârziere în dezvoltare care adesea nu este evidentă până la vârsta de aproximativ 6 luni și debutul ulterior al convulsiilor adesea greu de controlat, tremor, mers cu baze largi și ataxie cu un comportament vesel caracteristic (3). Există patru mecanisme moleculare recunoscute ale SA: deleții materne de novo ale cromozomului 15q11-q13 (70-80%); mutații ale genei UBE3A moștenite de la mamă (10-20%); disomie paternă 15 (3-5%); și defecte de amprentă (3-5%) în regiunea 15q11-q13 care modifică expresia genei UBE3A cauzale (21).
Indivizii cu SA nu sunt adesea observați de către cadrele medicale până la vârsta de ~6 luni, când sunt raportate întârzieri în dezvoltare, în special întârzieri în dezvoltarea motorie. Până în acest moment, părinții pot recunoaște comportamentul vesel care include râsul frecvent, zâmbetul și excitabilitatea. O scădere a nevoii de somn este raportată la >80% dintre persoanele cu SA (22). Aceștia dezvoltă adesea crize convulsive la vârste de 1-3 ani (23). Epilepsia poate fi intratabilă și are un aspect caracteristic pe EEG descris ca o putere delta crescută cu o undă trifazică caracteristică. Persoanele cu SA sunt descrise ca fiind ataxice în mișcările și mersul lor (24, 25). Microcefalia se poate dezvolta până la vârsta de ~2 ani. Comportamentele stereotipice includ dragostea pentru apă și hârtia încrețită, iar indivizii cu AS sunt caracteristic non-verbale și sunt categorisiți ca având un handicap intelectual sever. Cu toate acestea, este de remarcat faptul că indivizii cu SA au abilități care nu sunt bine surprinse de testele neuropsihologice obiective disponibile în prezent. Aceștia au abilități puternice în manipularea dispozitivelor electronice, dar comportamentele pot fi dificile și includ anxietate cu o durată scurtă de atenție.
Deoarece pacienții cu SPW sau AS pot prezenta fenotipuri variabile în funcție de clasa moleculară și pentru că există abordări potențiale de tratament și supraveghere pentru fiecare dintre acestea, este necesară o organigramă logică pentru comandarea testelor genetice de către clinicianul care evaluează acești pacienți. Obiectivul raportului nostru este de a descrie constatările clinice și genetice ale acestor două tulburări de amprentă genomică și de a ilustra opțiunile de testare genetică disponibile în mediul clinic și ordinea în care diferitele teste genetice pot fi obținute în modul cel mai productiv.
Experiența geneticii de laborator în tulburările de amprentă a cromozomului 15
Sindromul Prader-Willi
Pentru a servi drept exemplu al importanței testării cu microarray SNP de înaltă rezoluție, a fost recrutată în SUA o cohortă mare multisite de 510 participanți cu PWS confirmat genetic și grupată în trei clase moleculare. Acestea au fost caracterizate în continuare ca subtipuri de deleție 15q11-q13, subclase de disomie maternă 15 și defecte ale centrului de amprentă (16). În această cea mai mare cohortă de PWS raportată, s-a constatat că 303 indivizi prezentau deleția 15q11-q13 (60 % din cazuri), compusă din 118 indivizi (38,9 %) care prezentau deleția tipică 15q11-q13 de tip I, mai mare, care implică punctele de ruptură BP1 și BP3 ale cromozomului 15q11-q13 și 165 de indivizi (54.5%) au avut deleția tipică mai mică 15q11-q13 de tip II care implică punctele de ruptură BP2 și BP3, 20 de indivizi având o deleție atipică mai mare sau mai mică decât deleția tipică 15q11-q13 (6,6%). La persoanele identificate ca având o deleție a cromozomului 15, este important să se ia în considerare dacă o translocație echilibrată ar putea fi prezentă la tatăl probandului, deoarece acest lucru crește riscul de recurență a SPW la descendenții tatălui. În ceea ce privește disomia maternă 15, 185 de persoane (36%) au prezentat disomie uniparentală maternă 15 (UPD15), 13 persoane (12,5%) având izodisomie totală a întregului cromozom 15 din cauza unor erori în meioza II maternă; 60 (57,7%) au prezentat izodisomie segmentară din cauza unor evenimente de încrucișare în meioza I maternă și 31 au prezentat heterodisomie (29,8%), în timp ce la 81 de persoane nu s-a efectuat o analiză SNP microarray și nu s-a determinat clasificarea disomiei 15 materne. În ceea ce privește defectele de amprentă din PWS, au fost găsiți 22 de indivizi (4%), 13 (76,5%) având un statut de epimutație fără deleție, patru indivizi (23,5%) aveau o microdeleție a centrului de amprentă, în timp ce la restul de cinci indivizi nu s-a stabilit un tip de defect de amprentă. Într-un studiu înrudit, Hartin et al. (8) au efectuat o analiză suplimentară a defectelor de amprentă în PWS folosind droplet digital PCR și secvențierea de generație următoare a întregului exom într-o cohortă separată de PWS de 15 pacienți neînrudiți și s-a constatat că doi indivizi sau 13% au avut un defect de microdeleție a centrului de amprentă. La cei 60 de indivizi cu izodisomie segmentară 15 raportată de Butler et al. (16), dimensiunea medie totală a pierderii de heterozigozitate (LOH) a fost de 25,1 Mb, cu un interval de 5-67,4 Mb și o dimensiune medie de 16,4 Mb pentru LOH-urile individuale. Treizeci și doi de indivizi au avut un singur segment LOH, 25 de indivizi au avut două segmente și trei indivizi au avut trei segmente. Cele mai frecvente situsuri LOH au fost regiunea proximală 15q11-q13 și regiunea distală 15q26, inclusiv benzile 15q12 și 15q26.1 ca fiind cele mai frecvent înregistrate.
Prezența determinării UPD15 materne și a subclasei specifice (izodisomie segmentară sau totală) poate avea un impact asupra diagnosticului și supravegherii îngrijirii medicale, deoarece o a doua afecțiune genetică poate fi prezentă dacă mama este purtătoare a unei alele genetice recesive localizate în regiunea LOH care conduce la două copii identice. Sute de gene potențial cauzatoare de boli se găsesc pe cromozomul 15, iar aceste boli ar trebui să fie verificate sau monitorizate îndeaproape la cei cu izodisomie segmentară sau totală a cromozomului 15. O propunere de organigramă a testelor genetice pentru identificarea diferitelor clase moleculare atât pentru pacienții cu PWS, cât și pentru cei cu AS poate fi văzută în Rezumatul grafic.
Sindromul Angelman
Au fost identificate patru clase moleculare recunoscute în AS, care pot fi clasificate în funcție de impactul asupra metilării regiunii cromozomului 15. Cel mai frecvent subtip este o deleție a 15q11 maternă.2-q13, așa cum se observă în mod similar în cazul PWS de origine paternă și se găsește la ~70% dintre persoanele cu SA (21). Cu toate acestea, în AS, deleția tipică de clasa II este mai frecventă. Această deleție tipică de clasă II, mai mică, are cel mai frecvent o dimensiune de aproximativ 5 Mb din BP2-BP3 și este prezentă în 50% din cazurile de AS cu deleție. Delețiile de clasa I au o dimensiune de 5-7 Mb și cuprind BP1-BP3 (40 % din cazurile de deleție). Delețiile atipice se pot extinde de la BP1 sau BP2-BP4 sau de la puncte de ruptură mai îndepărtate. La indivizii cu o deleție pe copia maternă a cromozomului 15, trebuie să se ia în considerare dacă există semne pe microarray-ul cromozomal care arată tulburări care indică faptul că ar putea exista o translocație maternă. Acest lucru crește riscul de recurență a SA la viitorii descendenți materni. Disomia paternă uniparentală 15 reprezintă 5-7% dintre persoanele cu SA. Defectele de amprentă reprezintă 3-5% din indivizii cu SA și sunt cauzate de defecte în centrul de control al amprentării, rezumate de Buiting et al. (26). La indivizii cu un defect în centrul de control al amprentei, marcarea epigenetică în linia germinală nu reușește să treacă în mod corespunzător de la un model paternal cu expresia UBE3A redusă la tăcere pentru a permite un model matern de expresie la nivelul genei UBE3A. În până la 50% din cazurile raportate, poate fi identificată o mutație în centrul de control al amprentei. Sunt raportate cazuri mozaice de defecte ale centrului de amprentă în care un procent de celule nu exprimă regiunea 15q11.2-q13 și pot fi mai frecvente decât se credea anterior (27). Ultimul defect genetic în AS nu are impact asupra rezultatelor testelor de metilare a ADN-ului, dar este cauzat de o mutație în gena UBE3A moștenită de la mamă. Mutațiile în această genă reprezintă 11% din cazurile de SA (28). O mutație UBE3A ar putea fi moștenită pe cale maternă și, prin urmare, este indicată efectuarea de teste țintite la mama pacientului pentru a exclude un risc de recurență de 50% la viitoarea sa descendență. Dacă se consideră că mutația este moștenită, recomandăm să se ia în considerare testarea bunicului matern al pacientei, deoarece acest lucru ar putea avea implicații pentru viitorii copii ai mătușii materne.
Discuție
Managementul medical al SPW și SA ar trebui să fie dirijat de o echipă multidisciplinară în timpul copilăriei. Atât sugarii cu SPW (mai frecvent), cât și cei cu SA pot prezenta insuficiență de creștere. Un dietetician joacă un rol important în îngrijire la început pentru a aborda eșecul de dezvoltare și mai târziu în copilărie pentru a evita obezitatea prin intervenția dietetică cu restricție și utilizarea programelor de exerciții fizice (care este o preocupare observată mai frecvent pentru PWS, dar acum recunoscută în AS la unele persoane). Sunt necesari geneticieni clinici, specialiști în ortopedie, medici de îngrijire primară, terapeuți ocupaționali (OT), fizicieni (PT) și logopedici (SLP) specializați, experți în sănătate mintală, specialiști în somn, experți în sănătate mintală și endocrinologi pentru a aborda multiplele probleme de sănătate care pot apărea în SPW. O echipă de AS include geneticieni clinicieni, neurologi, terapeuți specializați pentru servicii PT, OT și SLP, specialiști în somn, gastroenterologie, medicină fizică și de reabilitare, ortopedie și experți în sănătate mintală. Pentru PWS, îngrijirea medicală adecvată, managementul și consilierea sunt obiective pentru a controla creșterea în greutate și pentru a monitoriza și trata afecțiunile comorbide asociate, comportamentul și problemele psihiatrice. Deficiențele de creștere și alte deficiențe hormonale comune în această tulburare necesită tratament. Controlul riguros al dietei cu siguranță alimentară și un mediu de rutină gestionat cu exerciții fizice regulate sunt strategii importante pentru a controla hiperfagia, obezitatea și complicațiile aferente necesare pe tot parcursul vieții. SA necesită o intervenție timpurie, inclusiv cunoașterea intervențiilor terapeutice specializate, cum ar fi dispozitivele de comunicare augmentativă și asistivă și un program de întărire cu exerciții și activități intensive de dezvoltare pentru atingerea potențialului maxim (de exemplu, SPIDER), tratament timpuriu cu benzodiazepine pentru convulsii și terapie dietetică, cum ar fi utilizarea unei diete ketogenice. Maximizarea tuturor aspectelor de îngrijire, inclusiv a tulburărilor de somn și a constipației, influențează foarte mult controlul convulsiilor. Un centru specializat, familiarizat cu complexitatea și aspectele unice ale acestor tulburări, poate afecta rezultatul.
Diagnosticul precoce este vital pentru a asigura o intervenție timpurie atât pentru SPW, cât și pentru SA. Pentru PWS, un diagnostic timpuriu ar trebui făcut în timpul copilăriei pentru a iniția tratamentul cu hormon de creștere, pentru a gestiona preocupările legate de alimentație, obezitate, deficiențe hormonale, întârzieri în dezvoltare și probleme comportamentale. Diagnosticul în AS asigură, de asemenea, terapii timpurii care au impact asupra rezultatelor dezvoltării, precum și profilaxia crizelor, inclusiv pregătirea cu benzodiazepine adecvate. Alte intervenții care se pot dovedi benefice includ dietele specializate pentru persoanele cu SA, cum ar fi dieta ketogenică sau terapia cu indice glicemic scăzut (LGIT). Diagnosticul timpuriu poate, de asemenea, să scadă costurile îngrijirii medicale prin prevenirea spitalizărilor prelungite legate de problemele de alimentație la persoanele cu PWS și a convulsiilor la copiii cu AS.
Identificarea clasei moleculare PWS sau AS cu ajutorul testelor genetice avansate, cum ar fi microplăcile SNP de înaltă rezoluție, va permite un diagnostic mai precis, ceea ce va duce la indicatori mai buni pentru prognostic și la o consiliere genetică mai precisă a membrilor familiei. Microarray-urile SNP de înaltă rezoluție, analiza FISH, amplificarea cu sondă de ligatură specifică de metilare-multiplexă (MS-MLPA) și/sau genotiparea cromozomului 15 sunt toate utile în determinarea delețiilor 15q11-q13. Microplăcile SNP de înaltă rezoluție pot identifica subtipurile de deleție (tipice și atipice) atât în PWS, cât și în AS, precum și subclasele UPD15 (izodisomie segmentară și izodisomie totală). Subtipul de heterodisomie al UPD și defectele IC (microdeleție și epimutație), atât în PWS, cât și în AS, pot necesita o analiză suplimentară de diagnosticare, așa cum este ilustrat în Rezumatul grafic. Subtipul sau clasele au un impact asupra diagnosticului, a riscului potențial de recurență pentru membrii familiei, a prognosticului și a monitorizării altor afecțiuni genetice și a caracteristicilor de risc ridicat legate de clasa moleculară. De exemplu, trăsăturile autiste și psihoza sunt mai frecvente la cei cu PWS și disomie maternă 15 și pot fi legate de subclasele UPD15 specifice. Cei cu deleții mai mari din clasa I în AS sunt mai susceptibili de a dezvolta convulsii greu de tratat și microcefalie.
O diagramă de flux a testelor genetice care încorporează opțiunile de testare care sunt disponibile, inclusiv cele utilizate istoric atât pentru PWS, cât și pentru AS, sunt enumerate în Rezumatul grafic. Testarea pentru PWS sau AS începe adesea cu metilarea ADN-ului și, dacă este anormală, se trece apoi la alte metode de testare genetică, inclusiv microplăcile SNP de înaltă rezoluție sau testele MS-MLPA, pe baza disponibilității pentru clinicieni și familii în mediul lor clinic. De preferință, ar fi comandată o matrice SNP de înaltă rezoluție, care este disponibilă cu ușurință și în comerț în asistența medicală occidentalizată. Secvențierea de generație următoare (NGS) a exomului (sau a întregului genom) este, de asemenea, disponibilă pentru clinicieni, dar droplet digital PCR (ddPCR) este în prezent bazată pe cercetare (14). Matricile SNP pot identifica clase moleculare specifice la majoritatea pacienților care prezintă caracteristici de PWS (aproximativ 85% din cazuri) sau AS (aproximativ 80%), în timp ce restul pacienților vor avea nevoie de teste suplimentare, așa cum este descris în Rezumat grafic. Testele genetice avansate specifice (de exemplu, ddPCR) pot fi suficient de sensibile pentru a cuantifica mozaicismul și pot identifica un diagnostic la un subset mare de indivizi cu caracteristici clinice mai ușoare ale PWS și AS, dar sunt necesare mai multe cercetări.
Au fost găsite diferențe clinice timpurii atunci când s-au comparat cei cu PWS sau AS care au statusul de deleție față de cei fără deleție (29), inclusiv hipopigmentare la cei cu PWS și AS care au deleția 15q11-q13 (30). Ulterior, au fost raportate scoruri mai mari ale IQ-ului verbal (31) sau psihoze (32) la cei cu UPD15 maternă în comparație cu deleția la persoanele cu SPW. Mai mult, Butler și colab. (19) au raportat scoruri adaptative mai scăzute și mai multe comportamente obsesiv-compulsive la indivizii cu PWS cu deleția 15q11-q13 de tip I în comparație cu UPD15. Zarcone et al. (33) au raportat că indivizii cu PWS și deleția 15q11-q13 de tip I aveau mai multe compulsii cu privire la curățenia personală și comportamente compulsive care erau greu de întrerupt și care interferau cu activitățile sociale mai mult decât cei cu deleții de tip II sau UPD15. Într-un studiu de corelație fenotip-genotip în cazul sindromului Angelman, Moncla et al. (34) au raportat o activitate convulsivă crescută la cei cu deleție mai mare de clasa I în comparație cu cei fără deleție. Microcefalia, ataxia, hipotonia și dificultățile de hrănire sunt, de asemenea, mai probabile în subtipul cu deleție (3). Aceștia pot avea tulburări de limbaj mai severe, în special în ceea ce privește limbajul receptiv și trăsături autiste (21, 35). Indivizii cu SA cu UPD paternă pot avea un limbaj receptiv îmbunătățit, abilități motorii îmbunătățite și o prevalență redusă a crizelor convulsive. Indivizii mozaici pot avea, de asemenea, un fenotip mai blând, inclusiv abilități de limbaj îmbunătățite, funcționare adaptivă și mai puține convulsii (36).
Secvențierea exomului de generație următoare sau a întregului genom poate avea, de asemenea, un loc în evaluările genetice în SPW sau SA, în special la acei indivizi care prezintă constatări neobișnuite sau un diagnostic întârziat (de exemplu, izodisomie segmentată sau totală UPD15) și în cazurile în care ADN-ul parental nu este disponibil (8). Pentru a aborda utilizarea și tipul de teste genetice pentru PWS și AS, a fost elaborată o nouă organigramă a testelor genetice pentru clinician, așa cum este descrisă și ilustrată în Rezumat grafic. Această organigramă poate ajuta la comandarea testelor genetice pe baza prezentării clinice pentru a determina diagnosticul, managementul și tratamentul adecvat și pentru a furniza cele mai exacte informații de consiliere genetică pentru ceilalți membri ai familiei. Sugerăm utilizarea acestui algoritm pentru a finaliza definitiv diagnosticul atât pentru PWS, cât și pentru AS. Susținem că diagnosticul este incomplet fără cunoașterea subtipului genetic specific al pacientului pentru a ghida consilierea, orientarea anticipativă, managementul și opțiunile terapeutice probabile. Determinarea clasei moleculare este importantă pentru îngrijirea și tratamentul medical și utilă pentru clinicianul angajat în consilierea genetică a membrilor familiei pentru PWS sau AS.
Contribuții ale autorilor
MB și JD au contribuit la redactarea manuscrisului, la revizuirea literaturii de specialitate, au contribuit cu expertiza lor și au editat manuscrisul.
Finanțare
Recunoaștem grantul Institutului Național pentru Sănătatea Copilului și Dezvoltare Umană cu numărul HD02528.
Conflict de interese
Autorii declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
Recunoștințe
Recunoaștem recunoștința lui Grace Graham pentru expertiza pregătirii manuscrisului.
1. Bittel DC, Butler MG. Sindromul Prader-Willi: genetică clinică, citogenetică și biologie moleculară. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Managementul sindromului Prader-Willi. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | Full CrossRef Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Aspecte clinice și genetice ale sindromului Angelman. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Sindromul Prader-Willi. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | Full CrossRef Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Sindromul Prader-Willi: o revizuire a constatărilor clinice, genetice și endocrine. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | Reflect Full Text | Google Scholar
6. Butler MG. Cauze genetice unice și sindromice ale obezității: exemple ilustrative. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Imprinting genetic sugerat de heterodisomia maternă în sindromul Prader-Willi nondeleție. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | Full CrossRef Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analiza centrului de amprentă a sindromului Prader-Willi folosind droplet digital PCR și secvențierea de generație următoare a întregului exom. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | Text integral | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Trei frați cu sindromul Prader-Willi cauzat de microdeleții ale centrului de amprentare și revizuire. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | Refef Full Text | Google Scholar
10. Hassan M, Butler MG. Sindromul Prader-Willi și delețiile submicroscopice atipice 15q11-q13 cu sau fără defecte de amprentă. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Sindromul Prader-Willi și sindromul Schaaf-Yang: boli de neurodezvoltare care se intersectează la nivelul genei MAGEL2. Boli. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Fenotipul Prader-Willi cauzat de deficiența paternă pentru clusterul de ARN nucleolar mic HBII-85 C/D box. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | Full CrossRef Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Fenotip Prader-Willi-Like cauzat de o microdeleție atipică 15q11.2. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deleții ale cromozomului 15 ca o cauză a sindromului Prader-Willi. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | Full CrossRef Text | Google Scholar
15. Butler MG, Palmer CG. Originea parentală a deleției cromozomului 15 în sindromul Prader-Willi. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Clasificarea genetică moleculară în sindromul Prader-Willi: un studiu de cohortă în mai multe locuri. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | Full CrossRef Text | Google Scholar
17. Butler MG, Thompson T. Sindromul Prader-Willi: constatări clinice și genetice. Endocrinologist. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Sindromul Prader-Willi: înțelegerea actuală a cauzei și a diagnosticului. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Diferențe comportamentale între subiecții cu sindrom Prader-Willi și deleție de tip I sau II și disomie maternă. Pediatrie. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | Refef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Fazele nutriționale în sindromul Prader-Willi. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Fenotipurile distincte disting clasele moleculare ale sindromului Angelman. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | Full CrossRef Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Evaluarea prin mai multe metode a somnului la copiii cu sindromul Angelman: un studiu caz-controlat. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | Full CrossRef Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Cardul genetic de utilitate clinică pentru: Sindromul Angleman. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | Full CrossRef Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Comportament și manifestări neuropsihiatrice în sindromul Angelman. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | Full CrossRef Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Sindromul Angelman. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | Refef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Sindromul Angelman – perspective asupra unei tulburări neurogenetice rare. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | Ref. full text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Sindromul Angelman atipic datorat unui defect de amprentare mozaic: rapoarte de caz și revizuirea literaturii. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | Reflect Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | Refef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Studiu clinic și citogenic al 39 de persoane cu sindrom Prader-Labhart-Willi. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | Reflect Full Text | Google Scholar
30. Butler MG. Hipopigmentarea: o caracteristică comună a sindromului Prader-Labhart-Willi. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Caracteristicile intelectuale ale sindromului Prader-Willi: compararea subtipurilor genetice. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | PubMed Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Boală psihotică la persoanele cu sindromul Prader-Willi din cauza disomiei uniparentale materne a cromozomului 15. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. The relationship between compulsive behavior and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Corelația fenotip-genotip la 20 de pacienți cu sindrom Angelman cu deleție și 20 fără deleție. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Childers AK, Wheeler F, Duis J. Limbajul expresiv conservat ca determinant fenotipic al sindromului Angelman mozaic. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar