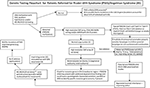
Graphical Abstract. Diagramma di flusso dei test genetici per i pazienti riferiti alla sindrome di Prader-Willi (PWS)/Sindrome di Angelman (AS). *Escludere traslocazioni o inversioni di Chr15 con studi cromosomici di routine; considerare altri disturbi genetici legati all’obesità; può richiedere lo screening del DNA della sindrome dell’X fragile per l’espansione della ripetizione del gene FMR1 o test genetici avanzati con sequenziamento di prossima generazione (NGS) per FMR1 o altre varianti di geni candidati utilizzando il sequenziamento dell’esoma intero (WES) o il sequenziamento dell’intero genoma (WGS; ad esempio, cause monogeniche di obesità). **Può essere usato per controllare lo stato di metilazione di altri geni imprinted Chr15; ddPCR, droplet digital PCR può essere usato per lo screening del mosaicismo.
Introduzione
I disordini del cromosoma 15 imprinting includono le sindromi di Prader-Willi (PWS) e Angelman (AS) (1-6) e duplicazioni del cromosoma 15q. La diagnosi di PWS o AS dipende dal genitore di origine e se l’espressione è aberrante limitata ai geni imprinting materni o paterni. La duplicazione 15q è causata da una copia aggiuntiva della regione 15q11.2-q13 di origine materna che può portare a convulsioni, problemi cognitivi e comportamentali tra cui il disturbo dello spettro autistico (ASD), ma non un fenotipo PWS o AS. La PWS deriva dalla perdita di geni maternamente impressi e paternamente espressi dalla regione del cromosoma 15q11-q13, mentre l’AS è causata dalla perdita di geni impressi e maternamente espressi in questa regione, con un impatto specifico sul gene UBE3A. A causa della natura imprinted dei geni responsabili, sia gli errori genetici che quelli epigenetici possono essere causativi.
Nel 1989, i soggetti con PWS e quelli senza delezione sono stati trovati con disomia 15 materna o con entrambi i 15 della madre quando si usavano marcatori polimorfici del DNA della regione prossimale 15q11-q13 (7). Più tardi, a metà degli anni ’90, lo sviluppo di sonde di DNA con ibridazione in situ fluorescente (FISH) è stato utilizzato per identificare le delezioni della regione 15q11-q13 sia nella PWS che nella SA. Il test di metilazione del DNA è stato sviluppato in questo periodo e un modello di metilazione anormale è stato visto in PWS e AS. Il test di metilazione del DNA è accurato al 99% nell’identificare la diagnosi di PWS, ma non identifica la classe molecolare individuale della PWS (2). Per l’AS, il test di metilazione del DNA identifica ~80% degli individui, ma di nuovo non distingue tra le classi molecolari o rileva una mutazione nel gene UBE3A che causa l’AS.
La tecnologia dei microarray è stata sviluppata all’inizio e alla metà degli anni 2000 e ha fatto avanzare la resa diagnostica. Ora i nuovi microarray SNP includono oltre due milioni di sonde di DNA e sono utili per individuare i sottotipi di delezione e le sottoclassi UPD15. Altre tecnologie come la droplet digital PCR (ddPCR) quantifica il numero di copie usando sonde di DNA del cromosoma 15 e può diagnosticare difetti genetici nella PWS o nella SA (8). Inoltre, i microarray SNP possono identificare LOHs definite come >8 Mb di dimensione e quando presenti sul cromosoma 15 supportano la diagnosi di disomia 15 materna o paterna in presenza di un pattern di metilazione del DNA anormale per PWS o AS, rispettivamente. La conferma del difetto di imprinting può richiedere non solo i microarray SNP per identificare le piccole microdelezioni, ma anche campioni di DNA dei genitori con genotipizzazione per identificare la presenza di un’eredità normale (biparentale) del cromosoma 15 che supporti la presenza di un difetto di imprinting di epimutazione nella PWS o nella SA, influenzando così i rischi di ricorrenza. La differenziazione di una microdelezione IC da uno stato di epimutazione non-delezione è clinicamente importante per le famiglie in quanto un rischio di recidiva del 50% è presente per ulteriori bambini se una microdelezione IC viene trovata nel genitore (9).
Ci sono più di una dozzina di geni e trascrizioni nella regione 15q11-q13 che sembrano avere un ruolo nella causalità della PWS e/o AS. I geni e le trascrizioni inclusi nell’area dai punti di rottura prossimali 15q11.2 BP1 e il punto di rottura distale 15q13 BP3 sono TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, RNA non codificanti (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 e HERC2. I geni MRKN3, MAGEL2, NDN, NIPAP1 e SNURF-SNRPN impressi sono espressi paternamente e quando sono disturbati possono causare caratteristiche della PWS. Per esempio, le mutazioni del gene MAGEL2 possono causare ipotonia neonatale, ritardo nello sviluppo, artrogriposi, caratteristiche autistiche, una scarsa aspirazione e obesità. Sono stati riportati anche pazienti con caratteristiche di PWS come risultato di piccole delezioni del trascritto non codificante SNORD116 (12) e altre delezioni simili nella regione (10, 13).
Ci siamo concentrati su AS e PWS in questo rapporto come entrambe le sindromi sono rilevate tramite test di metilazione del DNA, che permette di determinare l’allele genico parentale attivo e la diagnosi definitiva negli individui con PWS e nella maggior parte degli individui con AS (2). Tuttavia, il test di metilazione del DNA non identifica la classe molecolare in entrambe le sindromi. L’analisi cromosomica ad alta risoluzione è stata sviluppata e utilizzata nei primi anni ’80 ed è diventata un test standard di laboratorio basato sulla genetica per valutare la delezione del cromosoma 15q11-q13 identificata nella maggior parte dei pazienti con PWS a quel tempo (14) e successivamente per la SA. L’origine paterna della delezione 15q11-q13 è stata riportata nel 1983 (15) e trovata essere de novo, ma la dimensione della delezione 15q11-q13 o il tipo (tipico o atipico) non potevano essere determinati. Una diagnosi accurata e precoce con identificazione della classe molecolare è essenziale non solo per confermare la diagnosi clinica ma anche per la consulenza genetica, per informare la cura e il trattamento e per guidare le aspettative. Con l’intenzione di studi clinici in corso, una migliore comprensione dell’eziologia molecolare può avere un impatto sulle opportunità di partecipazione dei pazienti. Inoltre, gli studi imminenti includono oligonucleotidi antisenso per riattivare la copia paterna silenziata del cromosoma 15 negli individui con AS.
PWS e AS sono complessi disordini rari del neurosviluppo dovuti a errori di imprinting genomico. La PWS è riconosciuta come la causa genetica più comune di obesità pericolosa per la vita, se non controllata (2, 4, 6). Ci sono tre classi molecolari riconosciute di PWS che includono una delezione paterna 15q11-q13 di circa 5-6 Mb (60% dei casi) e la disomia materna 15 (UPD15) in cui entrambi i cromosomi 15 sono ereditati dalla madre (36%) che deriva dalla trisomia 15 con perdita del cromosoma 15 paterno all’inizio della gravidanza che porta a due cromosomi 15 dalla madre (16). La terza classe è un difetto del centro di imprinting. Se una microdelezione o un’epimutazione del centro di imprinting (IC), che controlla lo stato di espressione di alcuni geni imprintati sul cromosoma 15, è presente sull’allele paterno, si verifica la PWS. Questo difetto di imprinting è presente nel 4% degli individui con PWS (8, 16). La maggior parte dei casi di PWS sono sporadici con un’approssimativa equità tra gruppi etnici e sesso. La prevalenza stimata della PWS va da uno su 10.000 a uno su 30.000 (2). Il numero di individui affetti da PWS in tutto il mondo è stimato in circa 400.000 con circa 20.000 individui che vivono negli Stati Uniti (2, 17).
La PWS è caratterizzata da ipotonia infantile, scarso riflesso di suzione con difficoltà di alimentazione, bassa statura con mani e piedi piccoli, ipogonadismo secondario a carenze ormonali, lieve disabilità intellettuale, problemi di comportamento e iperfagia spesso con esordio tra i 6 e gli 8 anni di età che persiste in età adulta e porta all’obesità se non ci sono controlli ambientali. Durante l’infanzia, le caratteristiche craniofacciali sono visti tra cui un diametro stretto bifrontale, strabismo, piccolo naso rovesciato con un labbro superiore sottile, e gli angoli della bocca rovesciati, saliva appiccicosa, e smalto ipoplasia (2, 4, 6, 18). La cognizione è generalmente ridotta in base al background familiare e i problemi comportamentali che iniziano nell’infanzia includono l’autolesionismo (scaccolatura della pelle), gli scoppi, la testardaggine e gli scatti d’ira con problemi psichiatrici che si verificano durante questo periodo o più tardi nell’adolescenza o nella giovane età adulta (2). I problemi comportamentali includono ansia, disturbi dell’umore, psicosi e autismo che possono essere correlati a specifici sottotipi genetici di PWS o a classi molecolari (19).
Storicamente, la PWS è divisa in due fasi cliniche con l’incapacità di crescere durante l’infanzia che rappresenta la prima fase clinica e l’iperfagia con insorgenza di obesità che rappresenta la seconda fase (2). In seguito, le fasi nutrizionali sono state descritte per questo disordine genetico legato all’obesità e comprendono: Fase 0 con diminuzione del movimento fetale e ritardo della crescita in utero, seguita dalla Fase 1 relativa all’ipotonia, mancata crescita con difficoltà di alimentazione, Fase 2 che inizia a ~ 2 anni di età quando l’aumento di peso è notato per la prima volta e Fase 3 quando la mancanza di sazietà è accompagnata dalla ricerca di cibo e iperfagia che porta all’obesità, se non controllata esternamente. La fase 3 inizia intorno ai 6-8 anni di età (20).
La sindrome di Angelman è caratterizzata da un ritardo nello sviluppo spesso non evidente fino a circa 6 mesi di età e la successiva comparsa di crisi spesso difficili da controllare, tremore, andatura a base larga e atassia con un caratteristico contegno felice (3). Ci sono quattro meccanismi molecolari riconosciuti di AS: delezioni materne de novo del cromosoma 15q11-q13 (70-80%); mutazioni del gene UBE3A ereditato dalla madre (10-20%); disomia 15 paterna (3-5%); e difetti di imprinting (3-5%) all’interno della regione 15q11-q13 che alterano l’espressione del gene UBE3A causale (21).
Gli individui con AS spesso non vengono notati dai professionisti medici fino a circa 6 mesi di età, quando vengono riportati ritardi nello sviluppo, in particolare nello sviluppo motorio. A questo punto, i genitori possono riconoscere il comportamento felice che include frequenti risate, sorrisi ed eccitabilità. Un diminuito bisogno di sonno è riportato in >80% degli individui con AS (22). Spesso sviluppano crisi epilettiche all’età di 1-3 anni (23). L’epilessia può essere intrattabile e ha un aspetto caratteristico sull’EEG descritto come un aumento della potenza delta con una caratteristica onda trifasica. Gli individui con AS sono descritti come atassici nei loro movimenti e nel camminare (24, 25). La microcefalia può svilupparsi entro ~ 2 anni di età. I comportamenti stereotipati includono l’amore per l’acqua e la carta stropicciata e gli individui con AS sono caratteristicamente non verbali e classificati come gravemente disabili intellettuali. Tuttavia, è notevole che gli individui con AS hanno abilità non ben catturate sui test neuropsicologici oggettivi attualmente disponibili. Hanno forti abilità nella manipolazione dell’elettronica, ma i comportamenti possono essere impegnativi e comprendono l’ansia con brevi tempi di attenzione.
Come i pazienti con PWS o AS possono presentare fenotipi variabili a seconda della classe molecolare e perché esistono potenziali approcci di trattamento e sorveglianza per ciascuno, è necessario un diagramma di flusso logico per ordinare i test genetici da parte del clinico che valuta questi pazienti. L’obiettivo della nostra relazione è quello di descrivere i risultati clinici e genetici di questi due disturbi di imprinting genomico e illustrare le opzioni di test genetici disponibili nel contesto clinico e l’ordine in cui i diversi test genetici possono essere ottenuti in modo più produttivo.
Esperienza di genetica di laboratorio nei disturbi del cromosoma 15 Imprinting
Sindrome di Prader-Willi
Per servire da esempio dell’importanza dei test SNP microarray ad alta risoluzione, una grande coorte multisito di 510 partecipanti con PWS geneticamente confermata è stata reclutata negli USA e raggruppata in tre classi molecolari. Sono stati ulteriormente caratterizzati come sottotipi di delezione 15q11-q13, sottoclassi di disomia materna 15 e difetti del centro di imprinting (16). In questa più grande coorte PWS riportata, 303 individui sono stati trovati ad avere la delezione 15q11-q13 (60% dei casi) composta da 118 individui (38,9%) con la più grande delezione tipica 15q11-q13 di tipo I che coinvolge i breakpoint BP1 e BP3 del cromosoma 15q11-q13 e 165 individui (54.5%) avevano la più piccola delezione tipica 15q11-q13 di tipo II che coinvolge i breakpoints BP2 e BP3 con 20 individui che hanno una delezione atipica che è più grande o più piccola della delezione tipica 15q11-q13 (6,6%). Nelle persone identificate per avere una delezione del cromosoma 15, è importante considerare se una traslocazione bilanciata potrebbe essere presente nel padre del probando poiché questo aumenta il rischio di ricorrenza della PWS nella prole del padre. Per quanto riguarda la disomia materna 15, 185 individui (36%) avevano una disomia uniparentale materna 15 (UPD15) con 13 individui (12,5%) che avevano un’isodisomia totale dell’intero cromosoma 15 dovuta ad errori nella meiosi II materna; 60 (57,7%) mostravano un’isodisomia segmentale da eventi di crossover nella meiosi I materna e 31 mostravano un’eterodisomia (29,8%), mentre 81 individui non avevano un’analisi SNP microarray e la classificazione della disomia 15 materna determinata. Per quanto riguarda i difetti di imprinting della PWS, sono stati trovati 22 individui (4%) con 13 (76,5%) che avevano uno stato di epimutazione non-delezione, quattro individui (23,5%) avevano una microdelezione del centro di imprinting mentre gli altri cinque individui non avevano un tipo di difetto di imprinting stabilito. In uno studio correlato, un’ulteriore analisi dei difetti di imprinting nella PWS è stata effettuata da Hartin et al. (8) utilizzando la PCR digitale a goccia e il sequenziamento dell’esoma intero di prossima generazione in una coorte separata di PWS di 15 pazienti non imparentati e due individui o il 13% sono risultati avere un difetto di microdelezione del centro di imprinting. Nei 60 individui con isodisomia segmentale 15 riportati da Butler et al. (16), la dimensione media totale della perdita di eterozigosi (LOH) era di 25,1 Mb con un range di 5-67,4 Mb e una dimensione media di 16,4 Mb per le singole LOH. Trentadue individui avevano un segmento LOH, 25 individui avevano due segmenti e tre individui avevano tre segmenti. I siti di LOH più comuni erano la regione prossimale 15q11-q13 e la regione distale 15q26 che includeva le bande 15q12 e 15q26.1 come più comunemente registrate.
La presenza di UPD15 materna e la determinazione della sottoclasse specifica (isodisomia segmentale o totale) può avere un impatto sulla diagnosi e sulla sorveglianza medica in quanto una seconda condizione genetica può essere presente se la madre è portatrice di un allele genico recessivo situato nella regione LOH che porta a due copie identiche. Centinaia di geni potenzialmente causa di malattie si trovano sul cromosoma 15 e queste malattie dovrebbero essere controllate o monitorate attentamente in quelli con isodisomia segmentale o totale del cromosoma 15. Una proposta di diagramma di flusso dei test genetici per identificare le diverse classi molecolari per entrambi i pazienti PWS e AS può essere vista nel Graphical Abstract.
Sindrome di Angelman
Quattro classi molecolari riconosciute sono state identificate in AS che possono essere categorizzate dall’impatto sulla metilazione della regione del cromosoma 15. Il sottotipo più comune è una delezione del 15q11 materno.2-q13 di origine paterna nella PWS e si trova nel ~70% degli individui con AS (21). Tuttavia, nell’AS la tipica delezione di classe II è più comune. Questa tipica delezione di classe II, più piccola, ha una dimensione di circa 5 Mb da BP2-BP3 ed è presente nel 50% dei casi di delezione AS. Le delezioni di classe I hanno dimensioni di 5-7 Mb e comprendono BP1-BP3 (40% dei casi di delezione). Le delezioni atipiche possono estendersi da BP1 o BP2-BP4 o da breakpoint più distanti. Negli individui con una delezione sulla copia materna del cromosoma 15, si deve considerare se ci sono segni sul microarray cromosomico che mostrano disturbi che indicano che potrebbe esserci una traslocazione materna. Questo aumenta il rischio di ricorrenza dell’AS nella futura prole materna. La disomia uniparentale paterna 15 rappresenta il 5-7% degli individui con AS. I difetti di imprinting rappresentano il 3-5% degli individui con AS e sono causati da difetti nel centro di controllo dell’imprinting riassunti da Buiting et al. (26). Negli individui con un difetto nel centro di controllo dell’imprinting, la marcatura epigenetica nella linea germinale non riesce a passare correttamente da un modello paterno con espressione UBE3A silenziata per consentire un modello materno di espressione al gene UBE3A. Nel 50% dei casi riportati, può essere identificata una mutazione nel centro di controllo dell’imprinting. Casi mosaici di difetti del centro di imprinting in cui una percentuale di cellule manca l’espressione della regione 15q11.2-q13 è riportata e può essere più comune di quanto si pensasse in precedenza (27). L’ultimo difetto genetico nell’AS non influisce sui risultati dei test di metilazione del DNA, ma è causato da una mutazione nel gene UBE3A ereditato dalla madre. Le mutazioni in questo gene rappresentano l’11% dei casi di AS (28). Una mutazione UBE3A potrebbe essere ereditata dalla madre e quindi è indicato fare test mirati nella madre del paziente per escludere un rischio di recidiva del 50% nella sua futura prole. Se si ritiene che la mutazione sia ereditaria, raccomandiamo di prendere in considerazione l’esame del nonno materno del paziente, poiché questo potrebbe avere implicazioni per i futuri figli della zia materna.
Discussione
La gestione medica della PWS e della SA dovrebbe essere diretta da un team multidisciplinare durante l’infanzia. Sia i bambini con PWS (più comunemente) che quelli con AS possono avere problemi di crescita. Un dietista gioca un ruolo importante nell’assistenza all’inizio per affrontare l’insufficienza di crescita e più tardi nell’infanzia per evitare l’obesità con un intervento sulla dieta con restrizioni e l’uso di programmi di esercizio (che è una preoccupazione nota più comunemente per la PWS, ma ora riconosciuta nell’AS in alcuni individui). Genetisti clinici, specialisti ortopedici, medici di base, terapisti occupazionali (OT), fisici (PT) e del linguaggio (SLP) specializzati, esperti di salute mentale, specialisti del sonno, esperti di salute mentale ed endocrinologi sono necessari per affrontare i molteplici problemi di salute che possono verificarsi nella PWS. Un team AS include genetisti clinici, neurologi, terapisti specializzati per i servizi PT, OT e SLP, specialisti del sonno, gastroenterologia, medicina fisica e riabilitazione, ortopedia ed esperti di salute mentale. Per la PWS, cure mediche appropriate, gestione e consulenza sono obiettivi per controllare l’aumento di peso e per monitorare e trattare le condizioni comorbide associate, il comportamento e i problemi psichiatrici. Le carenze di crescita e di altri ormoni comuni in questo disturbo richiedono un trattamento. Un controllo rigoroso della dieta con sicurezza alimentare e un ambiente di routine gestito con regolare esercizio fisico sono strategie importanti per controllare l’iperfagia, l’obesità e le complicazioni correlate necessarie per tutta la vita. L’AS richiede un intervento precoce che includa la conoscenza di interventi terapeutici specializzati come i dispositivi di comunicazione aumentativa e assistiva e un programma di rafforzamento di esercizi e attività di sviluppo intensivo per raggiungere il massimo potenziale (ad esempio, SPIDER), il trattamento precoce con benzodiazepine per le crisi e la terapia dietetica come l’uso di una dieta chetogenica. Massimizzare tutti gli aspetti della cura, compresi i disturbi del sonno e la stitichezza, influenza notevolmente il controllo delle crisi. Un centro specializzato che abbia familiarità con le complessità e gli aspetti unici di questi disturbi può influenzare il risultato.
La diagnosi precoce è vitale per garantire un intervento tempestivo sia per la PWS che per l’AS. Per la PWS, una diagnosi precoce dovrebbe essere fatta durante l’infanzia per iniziare il trattamento con l’ormone della crescita, gestire i problemi di alimentazione, l’obesità, le carenze ormonali, i ritardi nello sviluppo e i problemi comportamentali. La diagnosi in AS assicura anche terapie precoci che hanno un impatto sui risultati dello sviluppo, così come la profilassi delle convulsioni, compresa la preparazione con benzodiazepine appropriate. Altri interventi che possono rivelarsi utili includono diete specializzate per gli individui con AS come la dieta chetogenica o la terapia a basso indice glicemico (LGIT). La diagnosi precoce può anche abbassare i costi delle cure mediche prevenendo lunghi ricoveri legati a problemi di alimentazione negli individui con PWS e convulsioni per i bambini con AS.
Identificare la classe molecolare PWS o AS con test genetici avanzati come i microarray SNP ad alta risoluzione permetterà una diagnosi più accurata, portando a migliori indicatori di prognosi, e una consulenza genetica più accurata dei membri della famiglia. I microarray SNP ad alta risoluzione, l’analisi FISH, l’amplificazione con sonda a legatura multipla specifica per la metilazione (MS-MLPA), e/o la genotipizzazione del cromosoma 15 sono tutti utili per determinare le delezioni 15q11-q13. I microarray SNP ad alta risoluzione possono identificare i sottotipi di delezione (tipici e atipici) sia in PWS che in AS, e le sottoclassi UPD15 (isodisomia segmentale e isodisomia totale). Il sottotipo di eterodisomia di UPD e i difetti IC (microdelezione ed epimutazione) sia nella PWS che nella SA possono richiedere un ulteriore lavoro diagnostico come illustrato nell’Abstract grafico. Il sottotipo o le classi influenzano la diagnosi, il potenziale rischio di ricorrenza per i membri della famiglia, la prognosi e il monitoraggio per altre condizioni genetiche e caratteristiche ad alto rischio legate alla classe molecolare. Per esempio, le caratteristiche autistiche e la psicosi sono più comuni in quelli con PWS e disomia 15 materna e possono essere correlate alle sottoclassi UPD15 specifiche. Quelli con le più grandi delezioni di classe I in AS hanno maggiori probabilità di sviluppare convulsioni difficili da trattare e microcefalia.
Un diagramma di flusso dei test genetici che incorpora le opzioni di test disponibili, compresi quelli usati storicamente sia per la PWS che per l’AS, sono elencati nell’Abstract grafico. I test per la PWS o la SA spesso iniziano con la metilazione del DNA e, se anormali, passano ad altri metodi di test genetici, compresi i microarray SNP ad alta risoluzione o i test MS-MLPA, in base alla disponibilità dei medici e delle famiglie nel loro ambiente clinico. Preferibilmente, verrebbe ordinato un array SNP ad alta risoluzione che è facilmente e commercialmente disponibile nelle cure mediche occidentali. Anche il sequenziamento di prossima generazione (NGS) dell’esoma (o dell’intero genoma) è disponibile per i medici, ma la droplet digital PCR (ddPCR) è attualmente basata sulla ricerca (14). Gli array SNP possono identificare specifiche classi molecolari nella maggior parte dei pazienti che presentano le caratteristiche della PWS (circa l’85% dei casi) o dell’AS (circa l’80%), mentre i pazienti rimanenti avranno bisogno di ulteriori test come descritto nel Graphical Abstract. Specifici test genetici avanzati (per esempio, ddPCR) possono essere adeguatamente sensibili per quantificare il mosaicismo e possono identificare una diagnosi in un ampio sottoinsieme di individui con caratteristiche cliniche più lievi di PWS e AS, ma sono necessarie ulteriori ricerche.
Sono state trovate differenze cliniche precoci quando si sono confrontati quelli con PWS o AS che avevano la delezione vs. lo stato di non-delezione (29) compresa l’ipopigmentazione in quelli con PWS e AS che avevano la delezione 15q11-q13 (30). In seguito, sono stati riportati punteggi più alti di QI verbale (31) o psicosi (32) in quelli con UPD15 materna rispetto alla delezione negli individui con PWS. Inoltre, Butler et al. (19) hanno riportato punteggi adattativi più bassi e più comportamenti ossessivo-compulsivi negli individui PWS con la delezione 15q11-q13 di tipo I rispetto alla UPD15. Zarcone et al. (33) hanno riferito che gli individui con PWS e la delezione 15q11-q13 di tipo I avevano più compulsioni con la pulizia personale e comportamenti compulsivi che erano difficili da interrompere e interferivano con le attività sociali più di quelli con delezioni di tipo II o UPD15. In uno studio di correlazione fenotipo-genotipo nella sindrome di Angelman, Moncla et al. (34) hanno riportato una maggiore attività convulsiva in quelli con la più grande delezione di tipo I rispetto a quelli senza delezione. Microcefalia, atassia, ipotonia e difficoltà di alimentazione sono anche più probabili nel sottotipo con delezione (3). Essi possono avere una più grave compromissione del linguaggio, in particolare del linguaggio ricettivo e dei tratti autistici (21, 35). Gli individui con AS con UPD paterna possono avere un miglior linguaggio ricettivo, migliori abilità motorie e una minore prevalenza di crisi. Gli individui con mosaico possono anche avere un fenotipo più lieve che include una migliore capacità di linguaggio, un funzionamento adattivo e meno crisi epilettiche (36).
Il sequenziamento dell’esoma o dell’intero genoma di nuova generazione può anche avere un posto nelle valutazioni genetiche nella PWS o AS, in particolare in quegli individui che presentano risultati insoliti o una diagnosi ritardata (ad esempio, UPD15 segmentale o isodisomia totale) e nei casi in cui il DNA dei genitori non è disponibile (8). Per affrontare l’uso e il tipo di test genetici per la PWS e la SA, è stato sviluppato un nuovo diagramma di flusso dei test genetici per il clinico, come descritto e illustrato nel Graphical Abstract. Questo diagramma di flusso può aiutare a ordinare i test genetici in base alla presentazione clinica per determinare la diagnosi, la gestione e il trattamento appropriati e per fornire le informazioni di consulenza genetica più accurate per gli altri membri della famiglia. Suggeriamo l’uso di questo algoritmo per completare definitivamente il lavoro diagnostico sia per la PWS che per l’AS. Noi sosteniamo che la diagnosi è incompleta senza la conoscenza del sottotipo genetico specifico del paziente per guidare la consulenza, la guida anticipata, la gestione e le probabili opzioni terapeutiche. La determinazione della classe molecolare è importante per l’assistenza e il trattamento medico e utile per il clinico impegnato nella consulenza genetica dei membri della famiglia per la PWS o l’AS.
Contributi degli autori
MB e JD hanno contribuito alla stesura del manoscritto, alla revisione della letteratura, hanno contribuito con la loro esperienza e hanno modificato il manoscritto.
Finanziamento
Riconosciamo il National Institute of Child Health and Human Development con la sovvenzione numero HD02528.
Conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Riconoscimenti
Riconosciamo Grace Graham per la preparazione del manoscritto.
1. Bittel DC, Butler MG. Sindrome di Prader-Willi: genetica clinica, citogenetica e biologia molecolare. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Gestione della sindrome di Prader-Willi. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Aspetti clinici e genetici della sindrome di Angelman. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Sindrome di Prader-Willi. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Sindrome di Prader-Willi: una revisione dei risultati clinici, genetici ed endocrini. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Singolo gene e cause sindromiche di obesità: esempi illustrativi. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Imprinting genetico suggerito da eterodisomia materna nella sindrome di Prader-Willi non delezione. Natura. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analisi del centro imprinting della sindrome di Prader-Willi utilizzando droplet digital PCR e next-generation whole-exome sequencing. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Tre fratelli con la sindrome di Prader-Willi causata da microdelezioni centro imprinting e recensione. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Sindrome di Prader-Willi e delezioni submicroscopiche atipiche 15q11-q13 con o senza difetti di imprinting. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fontana MD, Schaaf CP. La sindrome di Prader-Willi e la sindrome di Schaaf-Yang: malattie del neurosviluppo che si intersecano al gene MAGEL2. Malattie. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, tedesco JR, Shinawi M, Peters SU, Persona RE, et al. Prader-Willi fenotipo causato dalla carenza paterna per il cluster HBII-85 C/D box piccolo RNA nucleolare. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-Like fenotipo causato da un atipico 15q11.2 microdelezione. Geni (Basilea). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Delezioni del cromosoma 15 come causa della sindrome di Prader-Willi. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Origine parentale della delezione del cromosoma 15 nella sindrome di Prader-Willi. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Classificazione genetica molecolare nella sindrome di Prader-Willi: uno studio di coorte multisito. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Sindrome di Prader-Willi: risultati clinici e genetici. Endocrinologo. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Sindrome di Prader-Willi: comprensione attuale della causa e della diagnosi. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Differenze comportamentali tra i soggetti con sindrome di Prader-Willi e delezione di tipo I o II e disomia materna. Pediatria. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. fasi nutrizionali nella sindrome di Prader-Willi. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Fenotipi distinti distinguono le classi molecolari della sindrome di Angelman. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-metodo di valutazione del sonno nei bambini con sindrome di Angelman: uno studio caso-controllo. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. scheda gene utilità clinica per: Sindrome di Angleman. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Comportamento e manifestazioni neuropsichiatriche nella sindrome di Angelman. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. Una panoramica dei problemi di salute e lo sviluppo in una grande coorte clinica di bambini con sindrome di Angelman. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Sindrome di Angelman – intuizioni in un raro disturbo neurogenetico. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Sindrome di Angelman atipica a causa di un difetto di imprinting mosaico: case report e revisione della letteratura. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Sindrome di Angelman. Neuroterapeutica. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Indagine clinica e citogenica di 39 individui con la sindrome di Prader-Labhart-Willi. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Ipopigmentazione: una caratteristica comune della sindrome di Prader-Labhart-Willi. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Tetto E, pietra W, MacLean L, Feurer ID, Thompson T, Butler MG. Caratteristiche intellettuali della sindrome di Prader-Willi: confronto tra sottotipi genetici. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Malattia psicotica in persone con la sindrome di Prader-Willi a causa del cromosoma 15 disomia uniparentale materna. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. Il rapporto tra comportamento compulsivo e rendimento scolastico attraverso i tre sottotipi genetici della sindrome di Prader-Willi. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Correlazione fenotipo-genotipo in 20 delezione e 20 non delezione Angelman sindrome. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identificazione di nuove delezioni di 15q11q13 nella sindrome di Angelman da array-CGH: caratterizzazione molecolare e correlazioni genotipo-fenotipo. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Linguaggio espressivo conservato come un determinante fenotipico della sindrome di Angelman mosaico. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar