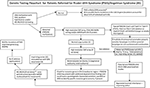
Résumé graphique. Organigramme des tests génétiques pour les patients adressés pour un syndrome de Prader-Willi (SPW)/syndrome d’Angelman (SA) . *Eliminer les translocations ou inversions de Chr15 par des études chromosomiques de routine ; envisager d’autres troubles génétiques liés à l’obésité ; peut nécessiter un dépistage ADN du syndrome du X fragile pour l’expansion de la répétition du gène FMR1 ou un test génétique avancé avec séquençage de nouvelle génération (NGS) pour FMR1 ou d’autres variantes de gènes candidats en utilisant le séquençage de l’exome entier (WES) ou le séquençage du génome entier (WGS ; par exemple, les causes monogéniques de l’obésité). **Peut être utilisé pour vérifier le statut de méthylation d’autres gènes imprimés Chr15 ; la ddPCR, la PCR numérique en gouttelettes peut être utilisée pour le dépistage du mosaïcisme.
Introduction
Les troubles de l’empreinte du chromosome 15 comprennent les syndromes de Prader-Willi (SPW) et d’Angelman (AS) (1-6) et les duplications du chromosome 15q. Le diagnostic du syndrome de Prader-Willi ou du syndrome d’Angelman dépend du parent d’origine et du fait que l’expression est limitée de manière aberrante aux gènes imprimés maternels ou paternels. La duplication 15q est causée par une copie supplémentaire de la région 15q11.2-q13 d’origine maternelle qui peut entraîner des crises d’épilepsie, des problèmes cognitifs et comportementaux, y compris des troubles du spectre autistique (TSA), mais pas un phénotype de SPW ou de SA. Le syndrome de Prader-Willi résulte de la perte de gènes imprimés et exprimés par la mère dans la région du chromosome 15q11-q13, tandis que le syndrome d’Autisme est causé par la perte de gènes imprimés et exprimés par la mère dans cette région, ce qui affecte spécifiquement le gène UBE3A. En raison de la nature imprégnée des gènes responsables, des erreurs génétiques et épigénétiques peuvent être causales.
En 1989, il a été constaté que les personnes atteintes du SPW et du statut de non-délétion présentaient une disomie maternelle 15 ou les deux 15 de la mère lors de l’utilisation de marqueurs ADN polymorphes de la région proximale 15q11-q13 (7). Plus tard, au milieu des années 1990, le développement de sondes d’ADN par hybridation in situ fluorescente (FISH) a permis d’identifier des délétions de la région 15q11-q13 dans le SPW et le SA. Les tests de méthylation de l’ADN ont été mis au point à cette époque et un schéma de méthylation anormal a été observé dans le SPW et le SA. Les tests de méthylation de l’ADN ont une précision d’environ 99 % pour le diagnostic du SPW, mais ne permettent pas d’identifier la classe moléculaire individuelle du SPW (2). Pour le SA, le test de méthylation de l’ADN identifie ~80% des individus, mais là encore, il ne permet pas de distinguer les classes moléculaires ou de détecter une mutation dans le gène UBE3A causant le SA.
La technologie des puces à ADN a été développée au début ou au milieu des années 2000 et a fait progresser le rendement diagnostique. Aujourd’hui, les nouvelles biopuces SNP comprennent plus de deux millions de sondes ADN et sont utiles pour détecter les sous-types de délétion et les sous-classes UPD15. D’autres technologies, comme la droplet digital PCR (ddPCR), quantifient le nombre de copies à l’aide de sondes d’ADN du chromosome 15 et peuvent diagnostiquer les anomalies génétiques dans le SPW ou le SA (8). De plus, les puces à SNP peuvent identifier les LOH définis comme étant de taille >8 Mb et, lorsqu’ils sont présents sur le chromosome 15, ils soutiennent le diagnostic de disomie 15 maternelle ou de disomie 15 paternelle en présence d’un schéma de méthylation anormal de l’ADN pour le SPW ou le SA, respectivement. La confirmation d’un défaut d’empreinte peut nécessiter non seulement des microréseaux SNP pour identifier les petites microdélétions, mais aussi des échantillons d’ADN parental avec génotypage pour identifier la présence d’une transmission normale (biparentale) du chromosome 15, ce qui confirme la présence d’un défaut d’empreinte par épimutation dans le cas du syndrome de Prader-Willi ou du syndrome d’Asperger, ce qui a un impact sur les risques de récurrence. La différenciation d’une microdélétion IC d’un statut d’épimutation de non-délétion est cliniquement importante pour les familles car un risque de récurrence de 50% est présent pour les enfants supplémentaires si une microdélétion IC est trouvée chez le parent (9).
Il existe plus d’une douzaine de gènes et de transcriptions dans la région 15q11-q13 qui semblent jouer un rôle dans la causalité du SPW et/ou du SA. Les gènes et les transcrits inclus dans la zone comprise entre les points de rupture proximale 15q11.2 BP1 et distale 15q13 BP3 sont TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, les ARN non codants (SNORD), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 et HERC2. Les gènes imprégnés MRKN3, MAGEL2, NDN, NIPAP1 et SNURF-SNRPN sont exprimés au niveau paternel et, lorsqu’ils sont perturbés, peuvent provoquer des caractéristiques du SPW. Par exemple, les mutations du gène MAGEL2 peuvent provoquer une hypotonie néonatale, un retard de développement, une arthrogrypose, des caractéristiques autistiques, une mauvaise succion et l’obésité . Des patients ont également été signalés avec des caractéristiques du SPW à la suite de petites délétions du transcrit non codant SNORD116 (12) et d’autres délétions similaires dans la région (10, 13).
Nous nous sommes concentrés sur la SA et le SPW dans ce rapport car les deux syndromes sont détectés par des tests de méthylation de l’ADN, qui permettent de déterminer l’allèle parental actif du gène et de poser un diagnostic définitif chez les personnes atteintes du SPW et chez la plupart des personnes atteintes de SA (2). Cependant, le test de méthylation de l’ADN ne permet pas d’identifier la classe moléculaire dans l’un ou l’autre des syndromes. L’analyse chromosomique à haute résolution a été développée et utilisée au début des années 1980 et est devenue un test génétique de laboratoire standard pour évaluer la délétion du chromosome 15q11-q13 identifiée chez la majorité des patients atteints du SPW à cette époque (14) et, plus tard, du SA. L’origine paternelle de la délétion 15q11-q13 a été signalée en 1983 (15) et s’est avérée être de novo, mais la taille de la délétion 15q11-q13 ou le type (typique ou atypique) n’a pas pu être déterminé. Un diagnostic précis et précoce avec identification de la classe moléculaire est essentiel non seulement pour confirmer le diagnostic clinique mais aussi pour le conseil génétique, pour informer les soins et le traitement et pour orienter les attentes. Compte tenu des essais cliniques en cours, une meilleure compréhension de l’étiologie moléculaire pourrait avoir un impact sur les possibilités de participation des patients. En outre, des essais imminents comprennent des oligonucléotides antisens pour réactiver la copie paternelle réduite au silence du chromosome 15 chez les personnes atteintes de SA.
Le SPW et la SA sont des troubles neurodéveloppementaux rares complexes dus à des erreurs d’empreinte génomique. Le SPW est reconnu comme la cause génétique la plus fréquente d’obésité potentiellement mortelle, si elle n’est pas contrôlée (2, 4, 6). Il existe trois classes moléculaires reconnues pour le syndrome de Prader-Willi, à savoir une délétion paternelle 15q11-q13 d’une taille d’environ 5-6 Mb (60 % des cas) et une disomie maternelle 15 (UPD15) dans laquelle les deux chromosomes 15 sont hérités de la mère (36 %), provenant d’une trisomie 15 avec perte du chromosome 15 paternel en début de grossesse, entraînant deux chromosomes 15 chez la mère (16). La troisième catégorie est une anomalie du centre d’empreinte. Si une microdélétion ou une épimutation du centre d’empreinte (CI), qui contrôle le statut d’expression de certains gènes imprimés sur le chromosome 15, est présente sur l’allèle paternel, le SPW apparaît. Ce défaut d’empreinte est observé chez 4 % des personnes atteintes du SPW (8, 16). La plupart des cas de SPW sont sporadiques avec une équité approximative entre les groupes ethniques et le sexe. La prévalence estimée du SPW est de un sur 10 000 à un sur 30 000 (2). Le nombre de personnes atteintes du SPW dans le monde serait de ~400 000, dont environ 20 000 aux États-Unis (2, 17).
Le SPW se caractérise par une hypotonie infantile, un mauvais réflexe de succion avec des difficultés d’alimentation, une petite taille avec des mains et des pieds de petite taille, un hypogonadisme secondaire à des déficiences hormonales, une légère déficience intellectuelle, des problèmes de comportement et une hyperphagie qui se manifeste souvent entre 6 et 8 ans et qui persiste à l’âge adulte et entraîne une obésité si aucun contrôle environnemental n’est mis en place. Pendant la petite enfance, on observe des traits cranio-faciaux caractéristiques, notamment un diamètre bifrontal étroit, un strabisme, un petit nez retroussé avec une lèvre supérieure mince et des coins de la bouche tournés vers le bas, une salive collante et une hypoplasie de l’émail (2, 4, 6, 18). La cognition est généralement réduite en fonction des antécédents familiaux et les problèmes de comportement qui commencent dans l’enfance comprennent l’automutilation (grattage de la peau), les crises, l’entêtement et les crises de colère, les problèmes psychiatriques survenant pendant cette période ou plus tard à l’adolescence ou au début de l’âge adulte (2). Les problèmes comportementaux comprennent l’anxiété, les troubles de l’humeur, la psychose et l’autisme qui peuvent être corrélés à des sous-types génétiques spécifiques du SPW ou à des classes moléculaires (19).
Historiquement, le SPW est divisé en deux stades cliniques, le retard de croissance pendant la petite enfance représentant le premier stade clinique et l’hyperphagie avec apparition de l’obésité représentant le deuxième stade (2). Par la suite, des phases nutritionnelles ont été décrites pour ce trouble génétique lié à l’obésité et comprennent : La phase 0 avec une diminution des mouvements du fœtus et un retard de croissance in utero, suivie de la phase 1 liée à l’hypotonie, au retard de croissance avec des difficultés d’alimentation, la phase 2 commençant à l’âge d’environ 2 ans lorsque la prise de poids est constatée pour la première fois et la phase 3 lorsque le manque de satiété s’accompagne d’une recherche de nourriture et d’une hyperphagie conduisant à l’obésité, si elle n’est pas contrôlée de l’extérieur. La phase 3 commence vers l’âge de 6 à 8 ans (20).
Le syndrome d’Angelman se caractérise par un retard de développement souvent non apparent jusqu’à l’âge de 6 mois environ et l’apparition ultérieure de crises souvent difficiles à contrôler, de tremblements, d’une démarche à base large et d’une ataxie avec un comportement heureux caractéristique (3). Il existe quatre mécanismes moléculaires reconnus de la SA : les délétions maternelles de novo du chromosome 15q11-q13 (70-80%) ; les mutations du gène UBE3A hérité maternellement (10-20%) ; la disomie 15 paternelle (3-5%) ; et les défauts d’empreinte (3-5%) dans la région 15q11-q13 qui modifient l’expression du gène UBE3A responsable (21).
Les individus atteints de SA ne sont souvent pas remarqués par les professionnels de la santé avant l’âge de ~6 mois, lorsque des retards de développement, en particulier un retard du développement moteur, sont signalés. À cette époque, les parents peuvent reconnaître le comportement joyeux qui comprend des rires fréquents, des sourires et de l’excitabilité. Une diminution du besoin de sommeil est signalée chez >80 % des personnes atteintes de SA (22). Ils développent souvent des crises à l’âge de 1 à 3 ans (23). L’épilepsie peut être réfractaire et a un aspect caractéristique sur l’EEG décrit comme une puissance delta accrue avec une onde triphasique caractéristique. Les personnes atteintes de SA sont décrites comme ataxiques dans leurs mouvements et leur marche (24, 25). La microcéphalie peut se développer vers l’âge de 2 ans. Les comportements stéréotypés comprennent l’amour de l’eau et du papier froissé. Les personnes atteintes de la SA sont généralement non verbales et classées dans la catégorie des déficiences intellectuelles sévères. Cependant, il convient de noter que les personnes atteintes de SA possèdent des aptitudes qui ne sont pas bien saisies par les tests neuropsychologiques objectifs actuellement disponibles. Ils ont de fortes capacités à manipuler l’électronique, mais les comportements peuvent être difficiles et inclure l’anxiété avec de courtes périodes d’attention.
Comme les patients atteints du SPW ou du SA peuvent présenter des phénotypes variables selon la classe moléculaire et parce qu’il existe des approches potentielles de traitement et de surveillance pour chacun, un organigramme logique est nécessaire pour la commande de tests génétiques par le clinicien qui évalue ces patients. L’objectif de notre rapport est de décrire les résultats cliniques et génétiques de ces deux troubles de l’empreinte génomique et d’illustrer les options de tests génétiques disponibles dans le cadre clinique et l’ordre dans lequel les différents tests génétiques peuvent être obtenus de la manière la plus productive.
Expérience de génétique de laboratoire dans les troubles de l’empreinte du chromosome 15
Syndrome de Prader-Willi
Pour servir d’exemple de l’importance des tests SNP microarray à haute résolution, une grande cohorte multisite de 510 participants présentant un SPW génétiquement confirmé a été recrutée aux États-Unis et regroupée en trois classes moléculaires. Elles ont été caractérisées en sous-types de délétion 15q11-q13, en sous-classes de disomie maternelle 15 et en défauts du centre d’empreinte (16). Dans cette cohorte la plus importante de cas de SPW, 303 personnes présentaient une délétion 15q11-q13 (60 % des cas), dont 118 (38,9 %) présentaient la plus grande délétion typique 15q11-q13 de type I impliquant les points de rupture BP1 et BP3 du chromosome 15q11-q13 et 165 personnes (54,5 %) présentaient la plus petite délétion typique 15q11-q13.Enfin, 20 personnes présentaient une délétion atypique plus grande ou plus petite que la délétion typique de 15q11-q13 (6,6 %). Chez les personnes identifiées comme ayant une délétion du chromosome 15, il est important de considérer si une translocation équilibrée pourrait être présente chez le père du proband, car cela augmente le risque de récurrence du SPW dans la descendance du père. En ce qui concerne la disomie maternelle 15, 185 individus (36 %) présentaient une disomie uniparentale maternelle 15 (UPD15), 13 individus (12,5 %) présentant une isodisomie totale de l’ensemble du chromosome 15 en raison d’erreurs dans la méiose II maternelle ; 60 (57,7 %) présentaient une isodisomie segmentaire due à des événements de croisement dans la méiose I maternelle et 31 présentaient une hétérodisomie (29,8 %), tandis que 81 individus n’ont pas fait l’objet d’une analyse de microréseau SNP et la classification de la disomie maternelle 15 n’a pas été déterminée. En ce qui concerne les défauts d’empreinte du syndrome de Prader-Willi, 22 individus (4 %) ont été trouvés, dont 13 (76,5 %) avaient un statut d’épimutation sans délétion, quatre individus (23,5 %) avaient une microdélétion du centre d’empreinte, tandis que les cinq autres individus n’avaient pas de type de défaut d’empreinte établi. Dans une étude connexe, une analyse plus approfondie des défauts d’empreinte dans le syndrome de Prader-Willi a été réalisée par Hartin et al. (8) à l’aide de la PCR numérique en gouttelettes et du séquençage de l’exome entier de nouvelle génération dans une cohorte distincte de 15 patients non apparentés atteints du syndrome de Prader-Willi ; deux individus, soit 13 %, présentaient un défaut de microdélétion du centre d’empreinte. Chez les 60 personnes atteintes d’isodisomie segmentaire 15 rapportées par Butler et al. (16), la taille moyenne totale de la perte d’hétérozygotie (LOH) était de 25,1 Mb, avec une plage de 5 à 67,4 Mb et une taille moyenne de 16,4 Mb pour les LOH individuelles. Trente-deux individus avaient un segment de LOH, 25 individus avaient deux segments et trois individus avaient trois segments. Les sites de LOH les plus fréquents étaient la région proximale 15q11-q13 et la région distale 15q26 comprenant les bandes 15q12 et 15q26.1 comme les plus couramment enregistrées.
La présence d’une UPD15 maternelle et la détermination de la sous-classe spécifique (isodisomie segmentaire ou totale) peuvent avoir un impact sur le diagnostic et la surveillance des soins médicaux car une deuxième condition génétique peut être présente si la mère est porteuse d’un allèle de gène récessif situé dans la région de LOH conduisant à deux copies identiques. Des centaines de gènes potentiellement pathogènes se trouvent sur le chromosome 15 et ces maladies doivent être vérifiées ou surveillées de près chez les personnes présentant une isodisomie segmentaire ou totale du chromosome 15. Un organigramme de test génétique proposé pour identifier les différentes classes moléculaires pour les patients atteints du SPW et du SA peut être vu dans le résumé graphique.
Syndrome d’Angelman
Quatre classes moléculaires reconnues ont été identifiées dans le SA qui peuvent être catégorisées par l’impact sur la méthylation de la région du chromosome 15. Le sous-type le plus courant est une délétion du 15q11 maternel.La délétion de la région 2-q13 est similaire à celle d’origine paternelle dans le SPW et se retrouve chez ~70% des individus atteints de SA (21). Cependant, dans le SA, la délétion typique de classe II est plus fréquente. Cette délétion typique de classe II, plus petite, a le plus souvent une taille d’environ 5 Mb entre BP2 et BP3 et est présente dans 50 % des cas de SA avec délétion. Les délétions de classe I ont une taille de 5 à 7 Mb et englobent BP1-BP3 (40 % des cas de délétion). Les délétions atypiques peuvent s’étendre à partir de BP1 ou BP2-BP4 ou de points de rupture plus éloignés. Chez les personnes présentant une délétion sur la copie maternelle du chromosome 15, il faut se demander s’il existe des signes sur le microréseau chromosomique montrant des perturbations qui indiquent qu’il pourrait y avoir une translocation maternelle. Cela augmente le risque de récurrence du SA dans la future descendance maternelle. La disomie paternelle uniparentale 15 représente 5 à 7 % des personnes atteintes de SA. Les défauts d’empreinte représentent 3 à 5 % des personnes atteintes de SA et sont causés par des défauts dans le centre de contrôle de l’empreinte, résumés par Buiting et al. (26). Chez les personnes présentant un défaut dans le centre de contrôle de l’empreinte, le marquage épigénétique dans la lignée germinale ne parvient pas à passer correctement d’un modèle paternel avec une expression réduite de l’UBE3A à un modèle maternel d’expression du gène UBE3A. Dans 50 % des cas signalés, une mutation dans le centre de contrôle de l’empreinte peut être identifiée. Des cas mosaïques de défauts du centre d’empreinte dans lesquels un pourcentage de cellules n’exprime pas la région 15q11.2-q13 sont signalés et pourraient être plus fréquents qu’on ne le pensait auparavant (27). La dernière anomalie génétique de la SA n’a pas d’incidence sur les résultats des tests de méthylation de l’ADN, mais elle est due à une mutation du gène UBE3A, hérité de la mère. Les mutations de ce gène sont responsables de 11 % des cas de SA (28). Une mutation du gène UBE3A pourrait être transmise par la mère et il est donc indiqué d’effectuer un test ciblé chez la mère du patient pour exclure un risque de récidive de 50 % dans sa future descendance. Si la mutation est jugée héréditaire, nous recommandons d’envisager de tester le grand-père maternel de la patiente car cela pourrait avoir des implications pour les futurs enfants de la tante maternelle.
Discussion
La prise en charge médicale du SPW et du SA doit être dirigée par une équipe multidisciplinaire pendant la petite enfance. Les nourrissons atteints de SPW (le plus souvent) et de SA peuvent tous deux présenter un retard de croissance. Un diététicien joue un rôle important dans la prise en charge, d’abord pour traiter le retard de croissance et, plus tard dans l’enfance, pour éviter l’obésité par une intervention diététique avec restriction et le recours à des programmes d’exercice (ce qui est une préoccupation constatée plus fréquemment pour le SPW, mais désormais reconnue dans le SA chez certains individus). Des généticiens cliniques, des spécialistes en orthopédie, des médecins de soins primaires, des ergothérapeutes (OT), des physiothérapeutes (PT) et des orthophonistes spécialisés, des experts en santé mentale, des spécialistes du sommeil, des experts en santé mentale et des endocrinologues sont nécessaires pour traiter les multiples problèmes de santé qui peuvent survenir dans le SPW. Une équipe de SA comprend des généticiens cliniciens, des neurologues, des thérapeutes spécialisés pour les services de PT, OT et SLP, des spécialistes du sommeil, de la gastro-entérologie, de la médecine physique et de la réadaptation, de l’orthopédie et des experts en santé mentale. Dans le cas du syndrome de Prader-Willi, les soins médicaux appropriés, la prise en charge et le conseil sont des objectifs pour contrôler la prise de poids et pour surveiller et traiter les conditions comorbides associées, les problèmes comportementaux et psychiatriques. Les déficiences en hormone de croissance et en autres hormones, fréquentes dans ce trouble, doivent être traitées. Un contrôle rigoureux du régime alimentaire avec une sécurité alimentaire et un environnement routinier géré avec de l’exercice régulier sont des stratégies importantes pour contrôler l’hyperphagie, l’obésité et les complications associées nécessaires tout au long de la vie. La SA nécessite une intervention précoce, y compris la connaissance des interventions thérapeutiques spécialisées telles que les dispositifs de communication améliorée et d’assistance et un programme de renforcement composé d’exercices et d’activités de développement intensif pour atteindre le potentiel maximal (par exemple, SPIDER), un traitement précoce avec des benzodiazépines pour les crises et une thérapie diététique telle que l’utilisation d’un régime cétogène. L’optimisation de tous les aspects des soins, y compris les troubles du sommeil et la constipation, influence grandement le contrôle des crises. Un centre spécialisé connaissant les subtilités et les aspects uniques de ces troubles peut influer sur le résultat.
Un diagnostic précoce est vital pour assurer une intervention précoce tant pour le SPW que pour le SA. Pour le SPW, un diagnostic précoce doit être posé pendant la petite enfance afin d’initier un traitement par hormone de croissance, de gérer les problèmes d’alimentation, l’obésité, les déficiences hormonales, les retards de développement et les problèmes de comportement. Le diagnostic de SA permet également de mettre en place des thérapies précoces qui ont un impact sur les résultats du développement, ainsi qu’une prophylaxie des crises, y compris une préparation avec des benzodiazépines appropriées. Parmi les autres interventions qui peuvent s’avérer bénéfiques, citons les régimes alimentaires spécialisés pour les personnes atteintes de SA, tels que le régime cétogène ou la thérapie à faible indice glycémique (LGIT). Un diagnostic précoce peut également réduire les coûts des soins médicaux en évitant les hospitalisations prolongées liées à des problèmes d’alimentation chez les personnes atteintes du SPW et les crises d’épilepsie chez les enfants atteints de SA.
L’identification de la classe moléculaire du SPW ou de la SA à l’aide de tests génétiques avancés tels que les microréseaux SNP à haute résolution permettra un diagnostic plus précis, conduisant à de meilleurs indicateurs de pronostic, et un conseil génétique plus précis des membres de la famille. Les puces SNP à haute résolution, l’analyse FISH, l’amplification par sonde de ligature multiplexée spécifique de la méthylation (MS-MLPA) et/ou le génotypage du chromosome 15 sont tous utiles pour déterminer les délétions 15q11-q13. Les puces SNP à haute résolution peuvent identifier les sous-types de délétions (typiques et atypiques) dans le SPW et le SA, ainsi que les sous-classes UPD15 (isodisomie segmentaire et isodisomie totale). Le sous-type d’hétérodisomie de l’UPD et les anomalies de l’IC (microdélétion et épimutation) dans le SPW et le SA peuvent nécessiter un bilan diagnostique supplémentaire, comme l’illustre le résumé graphique. Le sous-type ou les classes ont un impact sur le diagnostic, le risque potentiel de récurrence pour les membres de la famille, le pronostic et la surveillance d’autres maladies génétiques et des caractéristiques à haut risque liées à la classe moléculaire. Par exemple, les caractéristiques autistiques et la psychose sont plus fréquentes chez les personnes atteintes du syndrome de Prader-Willi et de la disomie 15 maternelle et peuvent être liées aux sous-classes spécifiques de l’UPD15. Ceux qui présentent les plus grandes délétions de classe I dans le SA sont plus susceptibles de développer des crises difficiles à traiter et une microcéphalie.
Un organigramme de tests génétiques incorporant les options de tests disponibles, y compris celles utilisées historiquement pour le SPW et le SA, est répertorié dans le résumé graphique. Le dépistage du syndrome de Prader-Willi ou du syndrome d’Asperger commence souvent par la méthylation de l’ADN et, en cas d’anomalie, passe ensuite à d’autres méthodes de dépistage génétique, y compris les microréseaux SNP à haute résolution ou les tests MS-MLPA, en fonction de la disponibilité pour les cliniciens et les familles dans leur environnement clinique. De préférence, on commandera une puce SNP à haute résolution qui est facilement disponible dans le commerce dans le cadre des soins médicaux occidentaux. Le séquençage de nouvelle génération (NGS) de l’exome (ou du génome entier) est également disponible pour les cliniciens, mais la PCR numérique en gouttelettes (ddPCR) fait actuellement l’objet de recherches (14). Les puces à ADN peuvent identifier des classes moléculaires spécifiques chez la majorité des patients présentant des caractéristiques du syndrome de Prader-Willi (environ 85 % des cas) ou du syndrome d’Asperger (environ 80 %), tandis que les autres patients devront subir des tests supplémentaires, comme décrit dans le résumé graphique. Des tests génétiques avancés spécifiques (par exemple, ddPCR) peuvent être suffisamment sensibles pour quantifier le mosaïcisme et peuvent identifier un diagnostic dans un large sous-ensemble de personnes présentant des caractéristiques cliniques plus légères du SPW et du SA, mais des recherches supplémentaires sont nécessaires.
Des différences cliniques précoces ont été trouvées en comparant les personnes atteintes du SPW ou du SA ayant le statut de délétion par rapport au statut de non-délétion (29), y compris une hypopigmentation chez les personnes atteintes du SPW et du SA ayant la délétion 15q11-q13 (30). Plus tard, des scores de QI verbal plus élevés (31) ou des psychoses (32) ont été signalés chez les personnes présentant une UPD15 maternelle par rapport à la délétion chez les personnes atteintes du SPW. En outre, Butler et al. (19) ont signalé des scores d’adaptation inférieurs et davantage de comportements obsessionnels-compulsifs chez les personnes atteintes du SPW présentant la délétion 15q11-q13 de type I par rapport à la délétion UPD15. Zarcone et al. (33) ont rapporté que les personnes atteintes du syndrome de Prader-Willi et présentant la délétion 15q11-q13 de type I avaient plus de compulsions liées à la propreté personnelle et un comportement compulsif difficile à interrompre et interférant avec les activités sociales que les personnes présentant les délétions de type II ou UPD15. Dans une étude de corrélation phénotype-génotype dans le syndrome d’Angelman, Moncla et al. (34) ont signalé une augmentation de l’activité convulsive chez les personnes présentant la plus grande délétion de classe I par rapport aux personnes sans délétion. La microcéphalie, l’ataxie, l’hypotonie et les difficultés d’alimentation sont également plus probables dans le sous-type avec délétion (3). Ils peuvent présenter des troubles du langage plus sévères, en particulier du langage réceptif, et des traits autistiques (21, 35). Les personnes atteintes de SA avec UPD paternelle peuvent avoir un meilleur langage réceptif, de meilleures capacités motrices et une prévalence réduite des crises. Les individus mosaïques peuvent également présenter un phénotype plus léger, y compris de meilleures capacités linguistiques, un meilleur fonctionnement adaptatif et moins de crises (36).
Le séquençage de l’exome ou du génome entier de nouvelle génération peut également avoir une place dans les évaluations génétiques du SPW ou du SA, en particulier chez les individus présentant des résultats inhabituels ou un retard de diagnostic (par exemple, isodisomie segmentaire ou totale UPD15) et dans les cas où l’ADN parental n’est pas disponible (8). Afin d’aborder l’utilisation et le type de tests génétiques pour le SPW et le SA, un nouvel organigramme de tests génétiques a été élaboré pour le clinicien, tel que décrit et illustré dans le résumé graphique. Cet organigramme peut aider à commander des tests génétiques en fonction de la présentation clinique afin de déterminer le diagnostic, la prise en charge et le traitement appropriés et de fournir les informations les plus précises en matière de conseil génétique aux autres membres de la famille. Nous suggérons l’utilisation de cet algorithme pour compléter définitivement le bilan diagnostique du SPW et du SA. Nous soutenons que le diagnostic est incomplet si l’on ne connaît pas le sous-type génétique spécifique du patient pour orienter le conseil, l’orientation anticipée, la prise en charge et les options thérapeutiques probables. La détermination de la classe moléculaire est importante pour les soins médicaux et le traitement et utile pour le clinicien engagé dans le conseil génétique des membres de la famille pour le SPW ou le SA.
Contributions des auteurs
MB et JD ont contribué à la rédaction du manuscrit, à la revue de la littérature, ont apporté leur expertise et ont édité le manuscrit.
Financement
Nous reconnaissons le numéro de subvention HD02528 de l’Institut national de la santé infantile et du développement humain.
Conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêts potentiel.
Remerciements
Nous remercions Grace Graham pour la préparation experte du manuscrit.
1. Bittel DC, Butler MG. Le syndrome de Prader-Willi : génétique clinique, cytogénétique et biologie moléculaire. Expert Rev Mol Med. (2005) 25:1-20. doi : 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Management of Prader-Willi Syndrome. New York, NY : Springer. (2006). doi : 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Aspects cliniques et génétiques du syndrome d’Angelman. Genet Med. (2010) 12:385-95. doi : 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Le syndrome de Prader-Willi. Genet Med. (2012) 14:10-26. doi : 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Le syndrome de Prader-Willi : une revue des résultats cliniques, génétiques et endocriniens. J Endocrinol Invest. (2015) 38:1249-63. doi : 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Causes monogéniques et syndromiques de l’obésité : exemples illustratifs. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi : 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi : 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analyse du centre d’empreinte du syndrome de Prader-Willi à l’aide de la PCR numérique en gouttelettes et du séquençage de l’exome entier de nouvelle génération. Mol Genet Genomic Med. (2019) 7:e00575. doi : 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Trois frères et sœurs atteints du syndrome de Prader-Willi causé par des microdélétions du centre d’imprégnation et revue. Am J Med Genet A. (2018) 176:886-95. doi : 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Syndrome de Prader-Willi et délétions submicroscopiques atypiques 15q11-q13 avec ou sans défauts d’empreinte. Eur J Med Genet. (2016) 59:584-9. doi : 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Syndrome de Prader-Willi et syndrome de Schaaf-Yang : maladies neurodéveloppementales se croisant au niveau du gène MAGEL2. Diseases. (2016) 13:4. doi : 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. (2008) 40:719-21. doi : 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-Like phenotype caused by an atypical 15q11.2 microdeletion. Genes (Bâle). (2020) 25:11. doi : 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. (1981) 304:325-9. doi : 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet. (1983) 4:1285-6. doi : 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Classification génétique moléculaire dans le syndrome de Prader-Willi : une étude de cohorte multisite. J Med Genet. (2019) 56:149-53. doi : 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Syndrome de Prader-Willi : résultats cliniques et génétiques. Endocrinologue. (2000) 10:3s-16s. doi : 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Le syndrome de Prader-Willi : compréhension actuelle de la cause et du diagnostic. Am J Med Genet. (1990) 35:319-32. doi : 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. (2004) 113:565-73. doi : 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. (2011) 155:1040-9. doi : 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Le syndrome d’Angelman. J Med Genet. (2001) 38:834-45. doi : 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome : a case-controlled study. Front Psychiatry. (2019) 10:874. doi : 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Carte génétique d’utilité clinique pour : Le syndrome d’Angleman. Eur J Hum Genet. (2015) 23:2. doi : 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Comportement et manifestations neuropsychiatriques dans le syndrome d’Angelman. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi : 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. (2020) 182:53-63. doi : 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelman syndrome – insights into a rare neurogenetic disorder. Nat Rev Neurol. (2016) 12:584-93. doi : 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Syndrome d’Angelman atypique dû à un défaut d’empreinte en mosaïque : rapports de cas et revue de la littérature. Am J Med Genet A. (2017) 173:753-7. doi : 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi : 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Étude clinique et cytogénique de 39 personnes atteintes du syndrome de Prader-Labhart-Willi. Am J Med Genet. (1986) 23:793-809. doi : 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hypopigmentation : une caractéristique commune du syndrome de Prader-Labhart-Willi. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Caractéristiques intellectuelles du syndrome de Prader-Willi : comparaison des sous-types génétiques. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi : 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. (2002) 359:135-6. doi : 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. The relationship between compulsive behavior and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intellect Disabil Res. (2007) 51:478-87. doi : 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. (1999) 7:131-9. doi : 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH : molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. (2007) 15:943-9. doi : 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preserved expressive language as a phenotypic determinant of mosaic Angelman syndrome. Mol Genet Genomic Med. (2019) 1:e837. doi : 10.1002/mgg3.837
CrossRef Full Text | Google Scholar
.