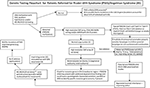
Grafisk sammanfattning. Flödesschema för genetisk testning för patienter som remitterats för Prader-Willis syndrom (PWS)/Angelmans syndrom (AS). * Uteslut Chr15-translokationer eller inversioner genom rutinmässiga kromosomstudier; överväg andra fetmarelaterade genetiska störningar; kan kräva DNA-screening för fragilt X-syndrom för expansion av FMR1-genens upprepning eller avancerad genetisk testning med nästa generations sekvensering (NGS) för FMR1 eller andra genvarianter som kan vara kandidater med hjälp av whole-exome-sekvensering (WES) eller whole-genome-sekvensering (WGS; t.ex. monogena orsaker till fetma). **Kan användas för att kontrollera metyleringsstatus för andra Chr15-präglade gener; ddPCR, droplet digital PCR kan användas för screening av mosaikism.
Introduktion
Kromosom 15-präglingsstörningar innefattar Prader-Willi- (PWS) och Angelman-syndromet (AS) (1-6) och kromosom 15q-duplikationer. Diagnosen av PWS eller AS beror på ursprungsföräldern och om uttrycket är avvikande begränsat till de maternella eller paternella präglade generna. Duplikation 15q orsakas av en extra kopia av den maternellt härledda 15q11.2-q13-regionen, vilket kan leda till anfall, kognitiva och beteendemässiga problem inklusive autismspektrumstörning (ASD), men inte till en PWS- eller AS-fenotyp. PWS uppstår på grund av förlust av maternellt präglade och paternellt uttryckta gener från kromosom 15q11-q13-regionen, medan AS orsakas av förlust av präglade och maternellt uttryckta gener i denna region, vilket särskilt påverkar UBE3A-genen. På grund av de ansvariga genernas präglade karaktär kan både genetiska och epigenetiska fel vara orsakande.
In 1989 konstaterades att de med både PWS och icke-deletionsstatus hade maternell disomi 15 eller båda 15:orna från modern när man använde polymorfa DNA-markörer från den proximala 15q11-q13-regionen (7). Senare, i mitten av 1990-talet, användes utvecklingen av fluorescerande in situ-hybridisering (FISH) DNA-prober för att identifiera deletioner av 15q11-q13-regionen i både PWS och AS. Metylerings-DNA-tester utvecklades under denna tidsperiod och ett onormalt metyleringsmönster sågs hos PWS och AS. Metylerings-DNA-testning är ~99 % exakt när det gäller att identifiera diagnosen PWS, men identifierar inte den enskilda PWS-molekylära klassen (2). För AS identifierar DNA-metyleringstest ~80 % av individerna, men skiljer återigen inte mellan de molekylära klasserna eller upptäcker en mutation i UBE3A-genen som orsakar AS.
Microarray-tekniken utvecklades i början och mitten av 2000-talet och har avancerat den diagnostiska avkastningen. Nu innehåller de nya SNP-mikroarrayerna över två miljoner DNA-sonder och är användbara för att upptäcka deletionssubtyper och UPD15-subklasser. Annan teknik som droplet digital PCR (ddPCR) kvantifierar antalet kopior med hjälp av DNA-sonder på kromosom 15 och kan diagnostisera genetiska defekter vid PWS eller AS (8). SNP-mikroarrayer kan dessutom identifiera LOH:er som definieras som >8 Mb stora och när de finns på kromosom 15 stödjer de diagnosen maternell disomi 15 eller paternell disomi 15 i närvaro av ett onormalt DNA-metyleringsmönster för PWS respektive AS. För att bekräfta en imprintingdefekt kan det krävas inte bara SNP-mikroarrayer för att identifiera små mikrodeletioner utan även DNA-prover från föräldrarna med genotypning för att identifiera förekomsten av normal (biparental) nedärvning av kromosom 15 som stödjer förekomsten av en epimutationsimprintingdefekt vid PWS eller AS, vilket påverkar risken för återfall. Differentiering av en IC-mikrodeletion från en icke-deletionsepimutationsstatus är kliniskt viktig för familjer eftersom det finns en 50-procentig återfallsrisk för ytterligare barn om en IC-mikrodeletion hittas hos föräldern (9).
Det finns över ett dussin gener och transkript i 15q11-q13-regionen som tycks spela en roll i orsakssambandet till PWS och/eller AS. Gener och transkript som ingår i området från den proximala 15q11.2-brytpunkten BP1 och den distala 15q13-brytpunkten BP3 är TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, icke-kodande RNA:er (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 och HERC2. De tryckta MRKN3-, MAGEL2-, NDN-, NIPAP1- och SNURF-SNRPN-generna uttrycks av fadern och när de är störda kan de ge upphov till PWS-egenskaper. Mutationer i MAGEL2-genen kan till exempel orsaka neonatal hypotoni, utvecklingsförsening, artrogrypos, autistiska drag, dålig sugförmåga och fetma . Patienter har också rapporterats med drag av PWS till följd av små deletioner av det icke-kodande SNORD116-transkriptet (12) och andra liknande deletioner i regionen (10, 13).
Vi fokuserade på AS och PWS i den här rapporten eftersom båda syndromen upptäcks via DNA-metyleringstester, vilket gör det möjligt att fastställa den aktiva föräldragenallelen och ställa en slutgiltig diagnos hos individer med PWS och hos de flesta individer med AS (2). DNA-metyleringstester identifierar dock inte den molekylära klassen i något av syndromen. Högupplöst kromosomanalys utvecklades och användes i början av 1980-talet och blev ett standardlaboratoriums genetiskt baserade test för att utvärdera den deletion av kromosom 15q11-q13 som då identifierades hos majoriteten av patienterna med PWS (14) och senare för AS. Det faderliga ursprunget till 15q11-q13-deletionen rapporterades 1983 (15) och konstaterades vara de novo, men storleken på 15q11-q13-deletionen eller typen (typisk vs. atypisk) kunde inte fastställas. En korrekt och tidig diagnos med identifiering av den molekylära klassen är viktig inte bara för att bekräfta den kliniska diagnosen utan också för genetisk rådgivning, för att informera om vård och behandling och för att vägleda förväntningarna. Med tanke på pågående kliniska prövningar kan en bättre förståelse av den molekylära etiologin påverka möjligheterna till patientdeltagande. Dessutom omfattar förestående försök antisense-oligonukleotider för att reaktivera den tystade faderliga kopian av kromosom 15 hos individer med AS.
PWS och AS är komplexa sällsynta neuroutvecklingsstörningar som beror på fel i den genomiska präglingen. PWS är erkänd som den vanligaste genetiska orsaken till livshotande fetma, om den lämnas okontrollerad (2, 4, 6). Det finns tre erkända molekylära klasser av PWS inklusive en paternell 15q11-q13-deletion som är ca 5-6 Mb stor (60 % av fallen) och maternell disomi 15 (UPD15) där båda kromosomerna 15 ärvs från modern (36 %) och som har sitt ursprung i trisomi 15 med förlust av faderns kromosom 15 under tidig graviditet, vilket leder till att modern har två kromosomer 15 (16). Den tredje klassen är en defekt i inpräglingscentret. Om en mikrodeletion eller epimutation av imprintingcentret (IC), som kontrollerar uttrycksstatusen för utvalda imprintinggener på kromosom 15, finns på den faderliga allelen uppstår PWS. Denna imprintingdefekt ses hos 4 % av individerna med PWS (8, 16). De flesta fall av PWS är sporadiska med en ungefärlig jämlikhet mellan etniska grupper och kön. Den uppskattade prevalensen av PWS är en på 10 000 till en på 30 000 (2). Antalet individer i världen med PWS tros vara ~400 000 med cirka 20 000 individer i USA (2, 17).
PWS kännetecknas av infantil hypotoni, dålig sugreflex med matningssvårigheter, kortväxthet med små händer och fötter, hypogonadism sekundärt till hormonbrist, lindrig intellektuell funktionsnedsättning, beteendeproblem och hyperfagi ofta med debut mellan 6 och 8 års ålder som kvarstår in i vuxen ålder och resulterar i fetma om miljökontroller inte finns på plats. Under spädbarnstiden ses karakteristiska kraniofaciala drag, bland annat en smal bifrontal diameter, strabism, liten uppåtvänd näsa med tunn överläpp och nedåtvända munhörn, klibbig saliv och emaljhypoplasi (2, 4, 6, 18). Kognitionen är i allmänhet nedsatt utifrån familjebakgrunden och beteendeproblem som börjar i barndomen inkluderar självskadebeteende (hudplockning), utbrott, envishet och humörsvängningar med psykiatriska problem som uppstår under denna tid eller senare i tonåren eller i unga vuxen ålder (2). Beteendeproblem inkluderar ångest, humörstörningar, psykos och autism som kan korrelera med specifika genetiska subtyper eller molekylära klasser av PWS (19).
Historiskt sett delas PWS in i två kliniska stadier där misslyckad tillväxt under spädbarnstiden representerar det första kliniska stadiet och hyperfagi med begynnande fetma representerar det andra stadiet (2). Senare har näringsfaser beskrivits för denna fetmarelaterade genetiska sjukdom och inkluderar: Fas 0 med minskade fosterrörelser och tillväxthämning i livmodern, följt av fas 1 med hypotoni, bristande utveckling med svårigheter att äta, fas 2 som börjar vid ~2 års ålder när viktökning först noteras och fas 3 när bristande mättnad åtföljs av födosökande och hyperfagi som leder till fetma, om den inte kontrolleras externt. Fas 3 börjar vid cirka 6-8 års ålder (20).
Angelmans syndrom kännetecknas av en utvecklingsförsening som ofta inte syns förrän vid cirka 6 månaders ålder och efterföljande debut av ofta svårkontrollerade anfall, tremor, bredbasig gång och ataxi med ett karakteristiskt glatt beteende (3). Det finns fyra erkända molekylära mekanismer för AS: de novo maternella deletioner av kromosom 15q11-q13 (70-80 %), mutationer av den maternellt nedärvda UBE3A-genen (10-20 %), paternell disomi 15 (3-5 %) och imprintingdefekter (3-5 %) inom 15q11-q13-regionen som förändrar uttrycket av den orsakande UBE3A-genen (21).
Individen med AS uppmärksammas ofta inte av sjukvårdspersonal förrän vid ~6 månaders ålder då förseningar i utvecklingen i synnerhet försenad motorisk utveckling rapporteras. Vid denna tidpunkt kan föräldrarna känna igen det glada beteendet som innefattar frekventa skratt, leenden och upphetsning. Ett minskat sömnbehov rapporteras hos >80 % av individerna med AS (22). De utvecklar ofta anfall i åldrarna 1-3 år (23). Epilepsi kan vara svårstoppad och har ett karakteristiskt utseende på EEG som beskrivs som en ökad deltakraft med en karakteristisk trifasisk våg. Individer med AS beskrivs som ataxiska i sina rörelser och i sin gång (24, 25). Mikrocefali kan utvecklas vid ~2 års ålder. Stereotypa beteenden inkluderar kärlek till vatten och skrynkligt papper och individer med AS är karakteristiskt icke verbala och kategoriseras som gravt intellektuellt funktionshindrade. Det är dock anmärkningsvärt att individer med AS har färdigheter som inte fångas väl upp av de för närvarande tillgängliga objektiva neuropsykologiska testerna. De har en stark förmåga att hantera elektronik, men beteendena kan vara utmanande och inbegripa ångest med kort uppmärksamhetsspann.
Då patienter med PWS eller AS kan uppvisa varierande fenotyper beroende på molekylärklass och eftersom det finns potentiella behandlings- och övervakningsmetoder för var och en av dem, behövs ett logiskt flödesschema för hur klinikern som utvärderar dessa patienter ska beställa genetiska tester. Fokus i vår rapport är att beskriva de kliniska och genetiska fynden för dessa två genomiska präglingsstörningar och illustrera de genetiska testalternativ som finns tillgängliga i den kliniska miljön och i vilken ordning de olika genetiska testerna kan erhållas på det mest produktiva sättet.
Laboratoriegenetisk erfarenhet av kromosom 15 Imprinting Disorders
Prader-Willis syndrom
För att tjäna som exempel på betydelsen av högupplöst SNP-mikroarray-testning rekryterades en stor multisite-kohort av 510 deltagare med genetiskt bekräftad PWS i USA och grupperades i tre molekylära klasser. De karaktäriserades vidare som 15q11-q13-deletionssubtyper, maternell disomi 15-subklasser och defekter i imprintingcenter (16). I denna största rapporterade PWS-kohort konstaterades 303 individer ha 15q11-q13-deletion (60 % av fallen) bestående av 118 individer (38,9 %) med den större typiska 15q11-q13-typ I-deletionen som involverar kromosom 15q11-q13-brytpunkterna BP1 och BP3 och 165 individer (54.5 %) hade den mindre typiska 15q11-q13 typ II-deletionen med brytpunkterna BP2 och BP3 och 20 individer hade en atypisk deletion som är större eller mindre än den typiska 15q11-q13-deletionen (6,6 %). Hos personer som identifierats ha en deletion av kromosom 15 är det viktigt att överväga om en balanserad translokation kan förekomma hos provpersonens far, eftersom detta ökar risken för återkommande PWS hos faderns avkomma. När det gäller moderlig disomi 15 hade 185 personer (36 %) moderlig uniparental disomi 15 (UPD15) med 13 personer (12,5 %) med total isodisomi av hela kromosom 15 på grund av fel i moderlig meios II; 60 (57,7 %) uppvisade segmentell isodisomi från crossover-händelser i moderlig meios I och 31 uppvisade heterodisomi (29,8 %), medan 81 personer inte hade någon SNP-mikroarray-analys och klassificering av moderlig disomi 15 fastställd. När det gäller PWS-imprintingdefekter hittades 22 individer (4 %) varav 13 (76,5 %) hade en icke-deletionsepimutationsstatus, fyra individer (23,5 %) hade en mikrodeletion av imprintingcentret medan de återstående fem individerna inte hade någon typ av imprintingdefekt fastställd. I en relaterad studie utförde Hartin et al. (8) ytterligare analyser av imprintingdefekter i PWS med hjälp av droplet digital PCR och nästa generations whole-exome-sekvensering i en separat PWS-kohort av 15 obesläktade patienter och två individer eller 13 % visade sig ha en mikrodeletionsdefekt i imprintingcentret. Hos de 60 individer med segmentell isodisomi 15 som rapporterades av Butler et al. (16) var den totala genomsnittliga storleken på förlust av heterozygositet (LOH) 25,1 Mb med ett intervall på 5-67,4 Mb och en genomsnittlig storlek på 16,4 Mb för enskilda LOH:er. Trettiotvå individer hade ett LOH-segment, 25 individer hade två segment och tre individer hade tre segment. De vanligaste LOH-platserna var den proximala 15q11-q13-regionen och den distala 15q26-regionen inklusive banden 15q12 och 15q26.1 som oftast registrerades.
Närvaron av maternell UPD15 och bestämning av specifik underklass (segmentell eller total isodisomi) kan påverka diagnosen och övervakningen av den medicinska vården, eftersom ett andra genetiskt tillstånd kan förekomma om mamman är bärare av en recessiv genallel som är belägen i LOH-regionen vilket leder till två identiska kopior. Hundratals potentiellt sjukdomsframkallande gener finns på kromosom 15 och dessa sjukdomar bör kontrolleras eller övervakas noga hos dem som har segmentell eller total isodisomi av kromosom 15. Ett förslag till flödesschema för genetisk testning för att identifiera de olika molekylära klasserna för både PWS- och AS-patienter kan ses i Graphical Abstract.
Angelmans syndrom
Fyra erkända molekylära klasser har identifierats i AS som kan kategoriseras efter inverkan på metyleringen av kromosom 15-regionen. Den vanligaste subtypen är en deletion av den maternella 15q11.2-q13-regionen som likaså ses av faderligt ursprung i PWS och finns hos ~70 % av individer med AS (21). I AS är dock den typiska klass II-deletionen vanligare. Denna typiska mindre klass II-deletion är oftast ungefär 5 Mb stor från BP2-BP3 och förekommer i 50 % av fallen med AS-deletion. Klass I-deletioner är 5-7 Mb stora och omfattar BP1-BP3 (40 % av fallen med deletion). Atypiska deletioner kan sträcka sig från BP1 eller BP2-BP4 eller från mer avlägsna brytpunkter. Hos individer med en deletion på den maternella kopian av kromosom 15 måste man överväga om det finns tecken på kromossomala mikroarray som visar störningar som tyder på att det kan finnas en maternell translokation. Detta ökar återfallsrisken för AS hos framtida moderns avkomma. Uniparental paternell disomi 15 står för 5-7 % av individerna med AS. Imprinting defekter står för 3-5 % av individerna med AS och orsakas av defekter i imprinting control center som sammanfattas av Buiting et al. (26). Hos individer med en defekt i imprinting-kontrollcentret misslyckas den epigenetiska markeringen i könscellerna med att korrekt växla från ett faderligt mönster med tystat UBE3A-uttryck för att möjliggöra ett moderligt uttrycksmönster för UBE3A-genen. I så många som 50 % av de rapporterade fallen kan en mutation i imprinting-kontrollcentret identifieras. Mosaikfall av defekter i imprintingcenter där en procentandel av cellerna saknar uttryck av 15q11.2-q13-regionen har rapporterats och kan vara vanligare än vad man tidigare trott (27). Den sista genetiska defekten i AS påverkar inte resultaten av DNA-metyleringstester, men orsakas av en mutation i den maternellt nedärvda UBE3A-genen. Mutationer i denna gen står för 11 % av AS-fallen (28). En UBE3A-mutation kan vara maternellt nedärvd och därför är det indicerat att göra riktade tester på patientens mor för att utesluta en 50-procentig återfallsrisk hos hennes framtida avkomma. Om mutationen bedöms vara ärftlig rekommenderar vi att man överväger att testa patientens morfar till modern eftersom detta kan få konsekvenser för moderns mosters framtida barn.
Diskussion
Den medicinska hanteringen av PWS och AS bör ledas av ett multidisciplinärt team under spädbarnstiden. Både spädbarn med PWS (vanligare) och AS kan få problem med att inte trivas. En dietist spelar en viktig roll i vården i början för att ta itu med störningar i utvecklingen och senare i barndomen för att undvika fetma med kostinterventioner med begränsning och användning av träningsprogram (vilket är ett problem som noterats oftare för PWS, men som nu erkänns i AS hos vissa individer). Kliniska genetiker, ortopedspecialister, primärvårdsläkare, specialiserade arbetsterapeuter (OT), fysioterapeuter (PT) och talterapeuter (SLP), experter på psykisk hälsa, sömnspecialister, experter på psykisk hälsa och endokrinologer behövs för att ta itu med de många olika hälsoproblem som kan uppstå i PWS. Ett AS-team omfattar kliniska genetiker, neurologer, specialiserade terapeuter för PT-, OT- och SLP-tjänster, sömnspecialister, gastroenterologi, fysisk medicin och rehabilitering, ortopedi och experter på mental hälsa. För PWS är lämplig medicinsk vård, hantering och rådgivning mål för att kontrollera viktökning och för att övervaka och behandla associerade komorbiditeter, beteende och psykiatriska problem. Brist på tillväxthormon och andra hormonbrister som är vanliga vid denna sjukdom kräver behandling. Rigorös kontroll av kosten med livsmedelssäkerhet och en hanterad rutinmässig miljö med regelbunden motion är viktiga strategier för att kontrollera hyperfagi, fetma och relaterade komplikationer som krävs under hela livet. AS kräver ett tidigt ingripande, inklusive kunskap om specialiserade terapeutiska interventioner, t.ex. förstärkande och stödjande kommunikationshjälpmedel och ett stärkande program med intensiva utvecklingsövningar och aktiviteter för att nå maximal potential (t.ex. SPIDER), tidig behandling med bensodiazepiner mot anfall och dietterapi, t.ex. användning av en ketogen diet. Maximering av alla aspekter av vården, inklusive sömnstörningar och förstoppning, påverkar i hög grad anfallskontrollen. Ett specialiserat center som är bekant med de invecklade och unika aspekterna av dessa störningar kan påverka resultatet.
En tidig diagnos är avgörande för att säkerställa ett tidigt ingripande för både PWS och AS. För PWS bör en tidig diagnos ställas under spädbarnstiden för att inleda behandling med tillväxthormon, hantera matningsproblem, fetma, hormonbrist, utvecklingsförseningar och beteendeproblem. Diagnosen vid AS säkerställer också tidiga terapier som påverkar utvecklingsresultaten samt anfallsprofylax inklusive förberedelse med lämpliga bensodiazepiner. Andra interventioner som kan visa sig vara fördelaktiga är bland annat specialiserade dieter för personer med AS, t.ex. ketogen kost eller behandling med lågt glykemiskt index (LGIT). Tidig diagnos kan också sänka kostnaderna för sjukvård genom att förhindra förlängda sjukhusvistelser relaterade till matningsproblem hos individer med PWS och kramper hos barn med AS.
Identifiering av den molekylära klassen för PWS eller AS med hjälp av avancerad genetisk testning, t.ex. högupplösta SNP-mikroarrayer, kommer att möjliggöra en mer exakt diagnos, vilket leder till bättre indikatorer för prognos och mer exakt genetisk rådgivning till familjemedlemmar. Högupplösta SNP-mikroarrays, FISH-analyser, metyleringsspecifik multipel ligeringssondamplifiering (MS-MLPA) och/eller genotypning av kromosom 15 är alla användbara för att fastställa 15q11-q13-deletioner. Högupplösta SNP-mikroarrays kan identifiera deletions-subtyper (typiska och atypiska) i både PWS och AS, och UPD15-subklasser (segmentell isodisomi och total isodisomi). Heterodisomi-subtypen av UPD och IC-defekter (mikrodeletion och epimutation) hos både PWS och AS kan kräva ytterligare diagnostik, vilket illustreras i det grafiska sammandraget. Subtypen eller klasserna påverkar diagnosen, potentiell återfallsrisk för familjemedlemmar, prognos och övervakning av andra genetiska tillstånd och högriskfunktioner i samband med den molekylära klassen. Exempelvis är autistiska drag och psykoser vanligare hos personer med PWS och moderlig disomi 15 och kan ha samband med de specifika UPD15-subklasserna. De med de större klass I-deletionerna i AS är mer benägna att utveckla svårbehandlade kramper och mikrocefali.
Ett flödesschema för genetisk testning med de testalternativ som finns tillgängliga, inklusive de som historiskt sett använts för både PWS och AS, finns förtecknat i Graphical Abstract. Testning för PWS eller AS börjar ofta med DNA-metylering och om det är onormalt går man sedan vidare till andra genetiska testmetoder, inklusive högupplösta SNP-mikroarrays eller MS-MLPA-analyser, baserat på tillgänglighet för kliniker och familjer i deras kliniska miljö. Företrädesvis beställs en högupplöst SNP-array som är lätt och kommersiellt tillgänglig inom västerländsk sjukvård. Nästa generations sekvensering (NGS) av exomet (eller hela genomet) är också tillgänglig för kliniker, men droplet digital PCR (ddPCR) är för närvarande forskningsbaserad (14). SNP-arrayer kan identifiera specifika molekylära klasser hos majoriteten av de patienter som uppvisar drag av PWS (cirka 85 % av fallen) eller AS (cirka 80 %) medan de återstående patienterna kommer att behöva ytterligare testning enligt beskrivningen i grafisk sammanfattning. Specifika avancerade genetiska tester (t.ex. ddPCR) kan vara tillräckligt känsliga för att kvantifiera mosaikism och kan identifiera en diagnos hos en stor delmängd individer med mildare kliniska drag av PWS och AS, men mer forskning behövs.
Främre kliniska skillnader hittades när man jämförde de personer med PWS eller AS som har deletion jämfört med icke-deletion (29), inklusive hypopigmentering hos de personer med PWS och AS som har 15q11-q13-deletion (30). Senare rapporterades högre verbala IQ-poäng (31) eller psykos (32) hos dem som hade moderns UPD15 jämfört med deletion hos individer med PWS. Vidare rapporterade Butler et al. (19) lägre adaptiva poäng och fler tvångsmässiga beteenden hos individer med PWS med 15q11-q13 typ I-deletion jämfört med UPD15. Zarcone et al. (33) rapporterade att personer med PWS och 15q11-q13 typ I-deletion hade fler tvångsbeteenden med personlig renlighet och tvångsbeteenden som var svåra att avbryta och som störde sociala aktiviteter mer än personer med typ II-deletion eller UPD15. I en fenotyp-genotyp korrelationsstudie av Angelmans syndrom rapporterade Moncla et al. (34) ökad anfallsaktivitet hos dem med den större typ I-deletionen jämfört med dem utan deletion. Mikrocefali, ataxi, hypotoni och matningssvårigheter är också mer sannolika i deletionssubtypen (3). De kan ha allvarligare språkstörningar i särskilt receptivt språk och autistiska drag (21, 35). Individer med AS med paternell UPD kan ha förbättrat receptivt språk, förbättrad motorisk förmåga och en minskad prevalens av anfall. Mosaikindivider kan också ha en mildare fenotyp, inklusive förbättrad språkförmåga, adaptiv funktion och färre anfall (36).
Nästa generationens exom- eller helgenomsekvensering kan också ha en plats i genetiska utvärderingar av PWS eller AS, särskilt hos de individer som uppvisar ovanliga fynd eller fördröjd diagnos (t.ex. UPD15-segmentell eller total isodisomi) och i de fall där föräldrarnas DNA inte är tillgängligt (8). För att ta itu med användningen och typen av genetisk testning för PWS och AS utvecklades ett nytt flödesschema för genetisk testning för klinikern som beskrivs och illustreras i Grafisk sammanfattning. Detta flödesschema kan vara till hjälp vid beställning av genetisk testning utifrån den kliniska presentationen för att fastställa lämplig diagnos, hantering och behandling och för att tillhandahålla den mest korrekta informationen om genetisk rådgivning till andra familjemedlemmar. Vi föreslår att denna algoritm används för att slutgiltigt slutföra det diagnostiska arbetet för både PWS och AS. Vi hävdar att diagnosen är ofullständig utan kunskap om patientens specifika genetiska subtyp för att vägleda rådgivningen, den föregripande vägledningen, hanteringen och de troliga terapeutiska alternativen. Den molekylära klassbestämningen är viktig för medicinsk vård och behandling och nyttig för den kliniker som är engagerad i genetisk rådgivning av familjemedlemmar för PWS eller AS.
Författarbidrag
MB och JD bidrog till att utforma manuskriptet, granska litteraturen, bidrog med sin expertis och redigerade manuskriptet.
Finansiering
Vi erkänner det nationella institutet för barnhälsa och mänsklig utveckling bidrag nummer HD02528.
Intressekonflikt
Författarna förklarar att forskningen har utförts i avsaknad av kommersiella eller finansiella relationer som skulle kunna tolkas som en potentiell intressekonflikt.
Acknowledgments
Vi tackar Grace Graham för expertförberedelse av manuskriptet.
1. Bittel DC, Butler MG. Prader-Willis syndrom: klinisk genetik, cytogenetik och molekylärbiologi. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Behandling av Prader-Willis syndrom. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Kliniska och genetiska aspekter av Angelmans syndrom. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willis syndrom. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Prader-Willis syndrom: en genomgång av kliniska, genetiska och endokrina fynd. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Enstaka gener och syndromiska orsaker till fetma: illustrativa exempel. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willis syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analys av Prader-Willis syndroms imprinting center med hjälp av droplet digital PCR och nästa generations whole-exome-sekvensering. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Tre syskon med Prader-Willis syndrom orsakade av mikrodeletioner i imprintingcenter och översikt. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Prader-Willis syndrom och atypiska submikroskopiska 15q11-q13-deletioner med eller utan imprintingdefekter. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Prader-Willis syndrom och Schaaf-Yangs syndrom: neuroutvecklingssjukdomar som korsar varandra vid MAGEL2-genen. Sjukdomar. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi-fenotyp orsakad av faderns brist på HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-liknande fenotyp orsakad av en atypisk 15q11.2-mikrodeletion. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deletioner av kromosom 15 som orsak till Prader-Willis syndrom. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Molekylärgenetisk klassificering av Prader-Willis syndrom: en kohortstudie på flera platser. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Prader-Willis syndrom: kliniska och genetiska fynd. Endokrinolog. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Prader-Willis syndrom: nuvarande förståelse av orsak och diagnos. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Angelmans syndrom. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a case-controlled study. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Klinisk nytta genkort för: Anglemans syndrom. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Beteende och neuropsykiatriska manifestationer vid Angelmans syndrom. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. En översikt över hälsofrågor och utveckling i en stor klinisk kohort av barn med Angelmans syndrom. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelmans syndrom – insikter om en sällsynt neurogenetisk sjukdom. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypiskt Angelmans syndrom på grund av en mosaisk imprintingdefekt: fallrapporter och litteraturgenomgång. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Klinisk och cytogen undersökning av 39 individer med Prader-Labhart-Willis syndrom. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.132023030307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hypopigmentering: ett vanligt inslag vid Prader-Labhart-Willis syndrom. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Intellektuella egenskaper hos Prader-Willis syndrom: jämförelse av genetiska subtyper. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psykotisk sjukdom hos personer med Prader-Willis syndrom på grund av kromosom 15 moderlig uniparental disomi. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. Förhållandet mellan tvångsbeteende och akademisk prestation i de tre genetiska subtyperna av Prader-Willis syndrom. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Korrelation mellan fenotyp och genotyp hos patienter med 20 deletion och 20 icke-deletion av Angelmans syndrom. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identifiering av nya deletioner av 15q11q13 i Angelmans syndrom genom array-CGH: molekylär karakterisering och genotyp-fenotypkorrelationer. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Bevarat expressivt språk som en fenotypisk bestämningsfaktor för mosaiskt Angelmans syndrom. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar