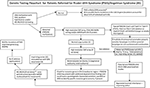
Graphical Abstract. Schemat postępowania w badaniach genetycznych u pacjentów kierowanych z rozpoznaniem zespołu Pradera-Williego (PWS)/Angelmana (AS). *Wykluczyć translokacje lub inwersje Chr15 za pomocą rutynowych badań chromosomów; rozważyć inne zaburzenia genetyczne związane z otyłością; może być konieczne przesiewowe badanie DNA w kierunku zespołu kruchego X w celu wykrycia ekspansji powtórzeń genu FMR1 lub zaawansowane badania genetyczne z sekwencjonowaniem następnej generacji (NGS) w kierunku FMR1 lub innych wariantów genów kandydujących za pomocą sekwencjonowania całego eksomu (WES) lub sekwencjonowania całego genomu (WGS; np. monogenowe przyczyny otyłości). **Może być stosowany do sprawdzenia statusu metylacji innych genów imprintingu Chr15; ddPCR, droplet digital PCR może być stosowany do badania mozaicyzmu.
Wprowadzenie
Zaburzenia imprintingu chromosomu 15 obejmują zespoły Pradera-Williego (PWS) i Angelmana (AS) (1-6) oraz duplikacje chromosomu 15q. Diagnoza PWS lub AS zależy od rodzica pochodzenia oraz od tego, czy ekspresja jest nieprawidłowo ograniczona do matczynych czy ojcowskich genów imprintingu. Duplikacja 15q jest spowodowana dodatkową kopią regionu 15q11.2-q13 pochodzącego od matki, co może prowadzić do napadów drgawkowych, problemów poznawczych i behawioralnych, w tym zaburzeń ze spektrum autyzmu (ASD), ale nie do fenotypu PWS lub AS. PWS wynika z utraty matczynie imprintowanych i ojcowsko wyrażonych genów z regionu chromosomu 15q11-q13, podczas gdy AS jest spowodowane utratą imprintowanych i matczynie wyrażonych genów w tym regionie, szczególnie wpływając na gen UBE3A. Ze względu na imprinted charakter odpowiedzialnych genów, zarówno genetyczne jak i epigenetyczne błędy mogą być przyczynowe.
W 1989 roku, osoby z PWS i statusem non-delecji zostały uznane za posiadające matczyną disomię 15 lub obie 15s od matki przy użyciu polimorficznych markerów DNA z proksymalnego regionu 15q11-q13 (7). Później, w połowie lat 90-tych, rozwój fluorescencyjnej hybrydyzacji in situ (FISH) sond DNA został wykorzystany do identyfikacji delecji regionu 15q11-q13 zarówno w PWS, jak i AS. W tym czasie opracowano badania metylacji DNA i zaobserwowano nieprawidłowy wzorzec metylacji w PWS i AS. Badanie metylacji DNA jest w ~99% dokładne w określaniu diagnozy PWS, ale nie identyfikuje indywidualnej klasy molekularnej PWS (2). W przypadku AS, badanie metylacji DNA identyfikuje ~80% osób, ale ponownie nie rozróżnia klas molekularnych lub nie wykrywa mutacji w genie UBE3A powodującej AS.
Technologia mikromacierzy została opracowana we wczesnych i średnich latach 2000 i posunęła naprzód wydajność diagnostyczną. Obecnie nowe mikromacierze SNP obejmują ponad dwa miliony sond DNA i są przydatne w wykrywaniu podtypów delecji i podklas UPD15. Inne technologie, takie jak kropelkowa cyfrowa PCR (ddPCR), określają liczbę kopii przy użyciu sond DNA chromosomu 15 i mogą diagnozować defekty genetyczne w PWS lub AS (8). Ponadto, mikromacierze SNP mogą zidentyfikować LOHs zdefiniowane jako >8 Mb w rozmiarze i jeśli są obecne na chromosomie 15, wspierają diagnozę matczynej disomii 15 lub ojcowskiej disomii 15 w obecności nieprawidłowego wzoru metylacji DNA odpowiednio dla PWS lub AS. Potwierdzenie defektu imprintingu może wymagać nie tylko mikromacierzy SNP w celu zidentyfikowania małych mikrodelecji, ale również próbek DNA rodziców z genotypowaniem w celu zidentyfikowania obecności normalnego (biparentalnego) dziedziczenia chromosomu 15s wspierającego obecność defektu imprintingu epimutacji w PWS lub AS, co wpływa na ryzyko nawrotów. Rozróżnienie mikrodelecji IC od statusu epimutacji bez delecji jest klinicznie ważne dla rodzin, ponieważ ryzyko nawrotu wynosi 50% dla kolejnych dzieci, jeśli mikrodelecja IC zostanie wykryta u rodzica (9).
Istnieje ponad tuzin genów i transkryptów w regionie 15q11-q13, które wydają się odgrywać rolę w wywoływaniu PWS i/lub AS. Geny i transkrypty zawarte w obszarze od proksymalnego 15q11.2 breakpoints BP1 i dystalnego 15q13 breakpoints BP3 to TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, niekodujące RNA (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 i HERC2. Imprintowane geny MRKN3, MAGEL2, NDN, NIPAP1 i SNURF-SNRPN ulegają ojcowskiej ekspresji i gdy są zaburzone mogą powodować cechy PWS. Na przykład, mutacje genu MAGEL2 mogą powodować hipotonię noworodkową, opóźnienie rozwoju, artrogrypozę, cechy autystyczne, słabe ssanie i otyłość. Opisano również pacjentów z cechami PWS w wyniku niewielkich delecji niekodującego transkryptu SNORD116 (12) i innych podobnych delecji w regionie (10, 13).
Skupiliśmy się na AS i PWS w tym raporcie, ponieważ oba zespoły są wykrywane poprzez badanie metylacji DNA, które pozwala na określenie aktywnego allelu genu rodzica i ostateczną diagnozę u osób z PWS i u większości osób z AS (2). Badanie metylacji DNA nie pozwala jednak na identyfikację klasy molekularnej w żadnym z tych zespołów. Analiza chromosomowa o wysokiej rozdzielczości została opracowana i zastosowana we wczesnych latach 80-tych i stała się standardowym laboratoryjnym testem genetycznym do oceny delecji chromosomu 15q11-q13 zidentyfikowanej u większości pacjentów z PWS w tym czasie (14), a później w przypadku AS. W 1983 roku doniesiono o ojcowskim pochodzeniu delecji 15q11-q13 (15) i stwierdzono, że jest to de novo, ale nie udało się określić wielkości delecji 15q11-q13 ani jej typu (typowa vs. atypowa). Dokładna i wczesna diagnoza z identyfikacją klasy molekularnej jest niezbędna nie tylko do potwierdzenia rozpoznania klinicznego, ale także do doradztwa genetycznego, informowania o opiece i leczeniu oraz kierowania oczekiwaniami. W związku z trwającymi badaniami klinicznymi, lepsze zrozumienie etiologii molekularnej może wpłynąć na możliwości udziału pacjentów w badaniach. Ponadto, zbliżające się próby obejmują oligonukleotydy antysensowe w celu reaktywacji wyciszonej ojcowskiej kopii chromosomu 15 u osób z AS.
PWS i AS są złożonymi, rzadkimi zaburzeniami neurorozwojowymi spowodowanymi błędami w imprintingu genomowym. PWS jest uznawana za najczęstszą genetyczną przyczynę zagrażającej życiu otyłości, jeśli pozostaje niekontrolowana (2, 4, 6). Istnieją trzy uznane klasy molekularne PWS, w tym ojcowska delecja 15q11-q13 o wielkości około 5-6 Mb (60% przypadków) oraz matczyna dysomia 15 (UPD15), w której oba chromosomy 15 są dziedziczone od matki (36%) pochodząca z trisomii 15 z utratą ojcowskiego chromosomu 15 we wczesnej ciąży prowadzącą do dwóch chromosomów 15 od matki (16). Trzecią klasą jest defekt centrum imprintingu. Jeżeli mikrodelecja lub epimutacja centrum imprintingu (IC), które kontroluje status ekspresji wybranych genów imprintingu na chromosomie 15, jest obecna na allelu ojcowskim, wówczas występuje PWS. Ten defekt imprintingu obserwowany jest u 4% osób z PWS (8, 16). Większość przypadków PWS ma charakter sporadyczny, z przybliżoną równomiernością pomiędzy grupami etnicznymi i płcią. Szacuje się, że częstość występowania PWS wynosi od 1 na 10 000 do 1 na 30 000 (2). Uważa się, że liczba osób na świecie z PWS wynosi ~400,000, z około 20,000 osób żyjących w USA (2, 17).
PWS charakteryzuje się niemowlęcą hipotonią, słabym odruchem ssania z trudnościami w karmieniu, niskim wzrostem z małymi dłońmi i stopami, hipogonadyzmem wtórnym do niedoborów hormonalnych, łagodną niepełnosprawnością intelektualną, problemami behawioralnymi i hiperfagią często z początkiem między 6 a 8 rokiem życia, która utrzymuje się w dorosłości i skutkuje otyłością, jeśli nie są stosowane kontrole środowiskowe. W okresie niemowlęcym obserwuje się charakterystyczne cechy czaszkowo-twarzowe, w tym wąską średnicę przedczołową, zeza, mały, odwrócony nos z cienką górną wargą i opadniętymi kącikami ust, lepką ślinę i hipoplazję szkliwa (2, 4, 6, 18). Problemy behawioralne rozpoczynające się w dzieciństwie obejmują samookaleczenia (skubanie skóry), wybuchy, upór i napady złości, a problemy psychiatryczne pojawiają się w tym czasie lub później w okresie dojrzewania lub w młodej dorosłości (2). Problemy behawioralne obejmują lęk, zaburzenia nastroju, psychozy i autyzm, które mogą korelować ze specyficznymi podtypami genetycznymi PWS lub klasami molekularnymi (19).
Historycznie, PWS jest podzielony na dwa etapy kliniczne z niepowodzeniem w rozwoju podczas niemowlęctwa reprezentujących pierwszy etap kliniczny i hiperfagii z początkiem otyłości reprezentujących drugi etap (2). Później, fazy żywieniowe zostały opisane dla tego związanego z otyłością zaburzenia genetycznego i obejmują: Fazę 0 ze zmniejszonymi ruchami płodu i opóźnieniem wzrostu in utero, a następnie Fazę 1 związaną z hipotonią, niepowodzeniem w rozwoju z trudnościami w karmieniu, Fazę 2 rozpoczynającą się w wieku ~2 lat, kiedy po raz pierwszy odnotowuje się przyrost masy ciała i Fazę 3, kiedy brakowi sytości towarzyszy poszukiwanie pokarmu i hiperfagia prowadząca do otyłości, jeśli nie jest zewnętrznie kontrolowana. Faza 3 rozpoczyna się w wieku około 6-8 lat (20).
Zespół Angelmana charakteryzuje się opóźnieniem rozwoju, które często nie jest widoczne do około 6 miesiąca życia, a następnie wystąpieniem często trudnych do opanowania napadów drżenia, drżenia, chodu o szerokim zakresie i ataksji z charakterystycznym radosnym usposobieniem (3). Istnieją cztery rozpoznane mechanizmy molekularne AS: de novo matczyne delecje chromosomu 15q11-q13 (70-80%); mutacje matczynego genu UBE3A (10-20%); ojcowska disomia 15 (3-5%); oraz defekty imprintingu (3-5%) w regionie 15q11-q13, które zmieniają ekspresję przyczynowego genu UBE3A (21).
Osoby z ZZSK często nie są zauważane przez pracowników służby zdrowia aż do ~6 miesiąca życia, kiedy zgłaszane są opóźnienia w rozwoju, w szczególności opóźnienie rozwoju motorycznego. Do tego czasu rodzice mogą rozpoznać radosne zachowanie, które obejmuje częsty śmiech, uśmiechanie się i pobudliwość. Zmniejszone zapotrzebowanie na sen zgłaszane jest u >80% osób z ZZSK (22). Często w wieku 1-3 lat pojawiają się u nich napady padaczkowe (23). Padaczka może być trudna do opanowania i ma charakterystyczny wygląd w EEG opisywany jako zwiększona moc delta z charakterystyczną falą trójfazową. Osoby z AS opisywane są jako ataksyjne w ruchach i chodzeniu (24, 25). Mikrocefalia może rozwinąć się do ~2 roku życia. Stereotypowe zachowania obejmują zamiłowanie do wody i marszczonego papieru, a osoby z ZZSK są charakterystycznie niewerbalne i klasyfikowane jako niepełnosprawne intelektualnie w stopniu znacznym. Warto jednak zauważyć, że osoby z AS posiadają umiejętności, które nie są dobrze uchwycone w obecnie dostępnych obiektywnych testach neuropsychologicznych. Mają duże zdolności manipulowania sprzętem elektronicznym, ale ich zachowania mogą być trudne i obejmować niepokój i krótki czas koncentracji uwagi.
Jako że pacjenci z PWS lub AS mogą prezentować zmienne fenotypy w zależności od klasy molekularnej i ponieważ dla każdego z nich istnieją potencjalne metody leczenia i nadzoru, potrzebny jest logiczny schemat postępowania przy zamawianiu badań genetycznych przez klinicystów oceniających tych pacjentów. Celem naszego doniesienia jest opisanie klinicznych i genetycznych wyników tych dwóch zaburzeń imprintingu genomowego oraz zilustrowanie opcji badań genetycznych dostępnych w warunkach klinicznych i kolejności, w jakiej różne badania genetyczne mogą być wykonywane w sposób najbardziej produktywny.
Doświadczenia genetyki laboratoryjnej w zaburzeniach imprintingu chromosomu 15
Zespół Bradera-Williego
Aby służyć jako przykład znaczenia badań mikromacierzy SNP o wysokiej rozdzielczości, w USA zrekrutowano dużą wielomiejscową kohortę 510 uczestników z genetycznie potwierdzonym PWS i pogrupowano ich w trzy klasy molekularne. Zostały one dalej scharakteryzowane jako podtypy delecji 15q11-q13, podklasy matczynej dysomii 15 i defekty centrum imprintingu (16). W tej największej kohorcie PWS u 303 osób stwierdzono delecję 15q11-q13 (60% przypadków), z czego 118 osób (38,9%) miało większą typową delecję 15q11-q13 typu I obejmującą punkty przerwania BP1 i BP3 chromosomu 15q11-q13, a 165 osób (54.5%) miało mniejszą typową delecję 15q11-q13 typu II obejmującą punkty przerwania BP2 i BP3, a 20 osób miało nietypową delecję, która jest większa lub mniejsza od typowej delecji 15q11-q13 (6,6%). U osób, u których stwierdzono delecję chromosomu 15, ważne jest rozważenie, czy translokacja zrównoważona może być obecna u ojca probanta, ponieważ zwiększa to ryzyko nawrotu PWS u potomstwa ojca. W przypadku matczynej dysomii 15, 185 osób (36%) miało matczyną dysomię jednorodzicielską 15 (UPD15), przy czym 13 osób (12,5%) miało całkowitą izodisomię całego chromosomu 15 z powodu błędów w matczynej mejozie II; 60 (57,7%) wykazywało segmentalną izodisomię spowodowaną zdarzeniami crossing-over w matczynej mejozie I, a 31 wykazywało heterodisomię (29,8%), podczas gdy u 81 osób nie przeprowadzono analizy mikromacierzy SNP i nie określono klasyfikacji matczynej dysomii 15. Jeśli chodzi o defekty imprintingu w PWS, stwierdzono je u 22 osób (4%), przy czym 13 (76,5%) miało status epimutacji niedelecyjnej, u czterech osób (23,5%) stwierdzono mikrodelecję centrum imprintingu, natomiast u pozostałych pięciu osób nie ustalono typu defektu imprintingu. W powiązanym badaniu, dalsza analiza defektów imprintingu w PWS została przeprowadzona przez Hartin i wsp. (8) przy użyciu kropelkowej cyfrowej PCR i sekwencjonowania całego eksomu następnej generacji w oddzielnej kohorcie PWS składającej się z 15 niespokrewnionych pacjentów i u dwóch osób lub 13% stwierdzono defekt mikrodelecji centrum imprintingu. W grupie 60 osób z segmentalną izodisomią 15 opisaną przez Butler i wsp. (16), całkowita średnia wielkość ubytku heterozygotyczności (LOH) wynosiła 25,1 Mb, przy zakresie 5-67,4 Mb i średniej wielkości 16,4 Mb dla poszczególnych LOH. Trzydzieści dwa osobniki miały jeden segment LOH, 25 osobników miało dwa segmenty, a trzy osobniki miały trzy segmenty. Najczęstszymi miejscami LOH były proksymalny region 15q11-q13 i dystalny region 15q26, w tym pasma 15q12 i 15q26.1 jako najczęściej rejestrowane.
Obecność matczynej UPD15 i specyficzne określenie podklasy (izodisomia segmentalna lub całkowita) może mieć wpływ na diagnozę i nadzór nad opieką medyczną, ponieważ druga choroba genetyczna może być obecna, jeśli matka jest nosicielem recesywnego allelu genu zlokalizowanego w regionie LOH, prowadzącego do powstania dwóch identycznych kopii. Na chromosomie 15 znajdują się setki genów potencjalnie powodujących choroby i choroby te powinny być ściśle kontrolowane lub monitorowane u osób z segmentalną lub całkowitą izodysomią chromosomu 15. Proponowany schemat badania genetycznego w celu identyfikacji różnych klas molekularnych zarówno dla pacjentów z PWS, jak i AS można zobaczyć w Graphical Abstract.
Zespół Angelmana
W AS zidentyfikowano cztery uznane klasy molekularne, które można podzielić na kategorie w zależności od wpływu na metylację regionu chromosomu 15. Najczęstszym podtypem jest delecja matczynego 15q11.region 2-q13, podobnie jak w PWS pochodzenia ojcowskiego i u ~70% osób z AS (21). Jednakże w AS typowa delecja klasy II jest bardziej powszechna. Ta typowa, mniejsza delecja klasy II najczęściej ma wielkość około 5 Mb od BP2-BP3 i jest obecna w 50% przypadków AS z delecją. Delecje klasy I mają wielkość 5-7 Mb i obejmują BP1-BP3 (40% przypadków delecji). Atypowe delecje mogą rozciągać się od BP1 lub BP2-BP4 lub od bardziej odległych punktów przerwania. U osób z delecją na matczynej kopii chromosomu 15 należy rozważyć, czy w mikromacierzy chromosomalnej nie występują zaburzenia wskazujące na możliwość translokacji matczynej. Zwiększa to ryzyko nawrotu AS u przyszłego potomstwa matki. Jednoparentalna disomia ojcowska 15 stanowi 5-7% osób z ZZSK. Defekty imprintingu stanowią 3-5% osób z AS i są spowodowane defektami w centrum kontroli imprintingu podsumowanymi przez Buitinga i wsp. (26). U osób z defektem w centrum kontroli imprintingu, znakowanie epigenetyczne w linii zarodkowej nie przełącza się prawidłowo z ojcowskiego wzorca z wyciszoną ekspresją UBE3A, aby umożliwić matczyny wzorzec ekspresji genu UBE3A. Aż w 50% opisanych przypadków można zidentyfikować mutację w centrum kontroli imprintingu. Opisywane są przypadki mozaikowe defektów centrum imprintingu, w których pewien odsetek komórek nie wykazuje ekspresji regionu 15q11.2-q13 i mogą one być częstsze niż wcześniej sądzono (27). Ostatni defekt genetyczny w ZZSK nie ma wpływu na wyniki badań metylacji DNA, ale jest spowodowany mutacją w dziedziczonym matczynie genie UBE3A. Mutacje w tym genie odpowiadają za 11% przypadków ZZSK (28). Mutacja UBE3A może być dziedziczona matczynie i dlatego wskazane jest wykonanie ukierunkowanych badań u matki pacjenta, aby wykluczyć 50% ryzyko nawrotu choroby u jej przyszłego potomstwa. Jeśli mutacja zostanie uznana za dziedziczną, zalecamy rozważenie przeprowadzenia badań u dziadka ze strony matki pacjentki, ponieważ może to mieć wpływ na przyszłe dzieci ciotki ze strony matki.
Dyskusja
Medyczne postępowanie w PWS i ZZSK powinno być kierowane przez wielodyscyplinarny zespół w okresie niemowlęcym. Zarówno niemowlęta z PWS (częściej), jak i z ZZSK mogą mieć problemy z przyrostem masy ciała. Dietetyk odgrywa ważną rolę w opiece nad niemowlętami z PWS, a w późniejszym dzieciństwie w zapobieganiu otyłości poprzez interwencje dietetyczne z ograniczeniem i wykorzystaniem programów ćwiczeń fizycznych (co jest problemem zauważalnym częściej w przypadku PWS, ale obecnie rozpoznawanym u niektórych osób z AS). Genetycy kliniczni, specjaliści ortopedzi, lekarze podstawowej opieki zdrowotnej, wyspecjalizowani terapeuci zajęciowi (OT), fizyczni (PT) i logopedzi (SLP), eksperci w dziedzinie zdrowia psychicznego, specjaliści w dziedzinie snu, eksperci w dziedzinie zdrowia psychicznego i endokrynolodzy są potrzebni, aby zająć się wieloma problemami zdrowotnymi, które mogą wystąpić w PWS. W skład zespołu AS wchodzą genetycy kliniczni, neurolodzy, wyspecjalizowani terapeuci PT, OT i SLP, specjaliści od snu, gastroenterolodzy, specjaliści medycyny fizycznej i rehabilitacji, ortopedzi oraz eksperci od zdrowia psychicznego. W przypadku PWS odpowiednia opieka medyczna, zarządzanie i doradztwo mają na celu kontrolę przyrostu masy ciała oraz monitorowanie i leczenie towarzyszących schorzeń, zachowań i problemów psychiatrycznych. Niedobory hormonów wzrostu i innych hormonów powszechne w tym zaburzeniu wymagają leczenia. Rygorystyczna kontrola diety z zabezpieczeniem żywnościowym i zarządzanie rutynowym środowiskiem z regularnymi ćwiczeniami są ważnymi strategiami kontroli hiperfagii, otyłości i związanych z nią powikłań wymaganych przez całe życie. ZZSK wymaga wczesnej interwencji obejmującej wiedzę na temat specjalistycznych interwencji terapeutycznych, takich jak wspomagające i wspomagające urządzenia komunikacyjne oraz program wzmacniający, obejmujący intensywne ćwiczenia rozwojowe i działania mające na celu osiągnięcie maksymalnego potencjału (np. SPIDER), wczesne leczenie benzodiazepinami napadów drgawkowych oraz dietoterapię, taką jak stosowanie diety ketogennej. Maksymalizacja wszystkich aspektów opieki, w tym zaburzeń snu i zaparć, ma duży wpływ na kontrolę napadów. Wyspecjalizowane centrum znające zawiłości i unikalne aspekty tych zaburzeń może wpłynąć na wynik leczenia.
Wczesna diagnoza jest niezbędna, aby zapewnić wczesną interwencję zarówno w przypadku PWS, jak i ZZSK. W przypadku PWS, wczesna diagnoza powinna być postawiona w okresie niemowlęcym, aby rozpocząć leczenie hormonem wzrostu, poradzić sobie z problemami żywieniowymi, otyłością, niedoborami hormonów, opóźnieniami rozwojowymi i problemami behawioralnymi. Diagnoza w ZZSK zapewnia również wczesną terapię, która ma wpływ na wyniki rozwojowe, a także profilaktykę napadów, w tym przygotowanie za pomocą odpowiednich benzodiazepin. Inne interwencje, które mogą okazać się korzystne, obejmują specjalistyczne diety dla osób z ZZSK, takie jak dieta ketogenna lub terapia niskim indeksem glikemicznym (LGIT). Wczesna diagnoza może również obniżyć koszty opieki medycznej poprzez zapobieganie przedłużonym hospitalizacjom związanym z problemami żywieniowymi u osób z PWS oraz napadom drgawkowym u dzieci z AS.
Identyfikacja klasy molekularnej PWS lub AS za pomocą zaawansowanych badań genetycznych, takich jak mikromacierze SNP o wysokiej rozdzielczości, pozwoli na dokładniejszą diagnozę, prowadząc do lepszych wskaźników rokowania oraz dokładniejszego poradnictwa genetycznego dla członków rodziny. Mikromacierze SNP o wysokiej rozdzielczości, analiza FISH, metylacja specyficzna dla multipleksowej amplifikacji sondy ligacyjnej (MS-MLPA) i/lub genotypowanie chromosomu 15 są przydatne w określaniu delecji 15q11-q13. Mikromacierze SNP o wysokiej rozdzielczości mogą zidentyfikować podtypy delecji (typowe i atypowe) zarówno w PWS, jak i w AS, oraz podklasy UPD15 (segmentalna izodisomia i całkowita izodisomia). Podtyp heterodisomii UPD oraz defekty IC (mikrodelecje i epimutacje) w PWS i AS mogą wymagać dodatkowej diagnostyki, jak pokazano na ilustracji graficznej. Podtyp lub klasy mają wpływ na diagnozę, potencjalne ryzyko nawrotu u członków rodziny, rokowanie i monitorowanie pod kątem innych chorób genetycznych oraz cech wysokiego ryzyka związanych z klasą molekularną. Na przykład, cechy autystyczne i psychozy występują częściej u osób z PWS i matczyną dysomią 15 i mogą być związane z określonymi podklasami UPD15. U osób z większymi delecjami klasy I w AS częściej występują trudne do leczenia napady drgawkowe i mikrocefalia.
Schemat blokowy badań genetycznych obejmujący dostępne opcje badań, w tym te stosowane historycznie zarówno w przypadku PWS, jak i AS, wymieniono w streszczeniu graficznym. Badania w kierunku PWS lub AS często rozpoczynają się od metylacji DNA i jeśli są nieprawidłowe, przechodzą do innych metod badań genetycznych, w tym mikromacierzy SNP o wysokiej rozdzielczości lub testów MS-MLPA, w zależności od dostępności dla klinicystów i rodzin w ich środowisku klinicznym. Najkorzystniej byłoby zamówić tablicę SNP o wysokiej rozdzielczości, która jest łatwo i komercyjnie dostępna w zachodniej opiece medycznej. Sekwencjonowanie następnej generacji (NGS) egzomu (lub całego genomu) jest również dostępne dla klinicystów, ale kropelkowa cyfrowa PCR (ddPCR) jest obecnie wykorzystywana w badaniach (14). Tablice SNP mogą zidentyfikować specyficzne klasy molekularne u większości pacjentów z cechami PWS (około 85% przypadków) lub AS (około 80%), podczas gdy pozostali pacjenci będą wymagali dodatkowych badań, jak opisano w abstrakcie graficznym. Specyficzne zaawansowane badania genetyczne (np. ddPCR) mogą być odpowiednio czułe do ilościowego określenia mozaicyzmu i mogą zidentyfikować diagnozę u dużej podgrupy osób z łagodniejszymi cechami klinicznymi PWS i AS, ale potrzebne są dalsze badania.
Wczesne różnice kliniczne stwierdzono porównując osoby z PWS lub AS z delecją i bez delecji (29), w tym hipopigmentację u osób z PWS i AS z delecją 15q11-q13 (30). W późniejszym okresie odnotowano wyższe wyniki werbalnego IQ (31) lub psychozy (32) u osób z matczyną UPD15 w porównaniu z delecją u osób z PWS. Co więcej, Butler i wsp. (19) odnotowali niższe wyniki adaptacyjne i więcej zachowań obsesyjno-kompulsywnych u osób z PWS z delecją 15q11-q13 typu I w porównaniu z UPD15. Zarcone i wsp. (33) donoszą, że osoby z PWS i delecją 15q11-q13 typu I mają więcej kompulsji związanych z czystością osobistą oraz zachowań kompulsywnych, które są trudne do przerwania i bardziej przeszkadzają w aktywności społecznej niż osoby z delecją typu II lub UPD15. W badaniu korelacji fenotyp-genotyp w zespole Angelmana, Moncla i wsp. (34) odnotowali zwiększoną aktywność napadową u osób z większą delecją klasy I w porównaniu z osobami bez delecji. Mikrocefalia, ataksja, hipotonia i trudności w karmieniu są również bardziej prawdopodobne w podtypie delecyjnym (3). Mogą występować u nich cięższe zaburzenia językowe, w szczególności języka receptywnego oraz cechy autystyczne (21, 35). Osoby z AS z ojcowskim UPD mogą mieć poprawiony język receptywny, poprawione zdolności motoryczne i zmniejszoną częstość występowania napadów. Osoby z mozaikowym UPD mogą mieć również łagodniejszy fenotyp, w tym lepsze zdolności językowe, funkcjonowanie adaptacyjne i mniejszą liczbę napadów (36).
Sekwencjonowanie egzomu lub całego genomu następnej generacji może mieć również swoje miejsce w ocenie genetycznej w PWS lub ZZSK, szczególnie u osób z nietypowymi wynikami lub opóźnioną diagnozą (np. segmentalna lub całkowita izodisomia UPD15) oraz w przypadkach, gdy DNA rodziców nie jest dostępne (8). Aby zająć się kwestią wykorzystania i rodzaju badań genetycznych w PWS i ZZSK, opracowano nowy schemat przeprowadzania badań genetycznych dla klinicystów, opisany i zilustrowany w streszczeniu graficznym. Schemat ten może być pomocny w zlecaniu badań genetycznych na podstawie obrazu klinicznego w celu ustalenia właściwego rozpoznania, postępowania i leczenia, a także w celu dostarczenia najdokładniejszych informacji dotyczących poradnictwa genetycznego dla pozostałych członków rodziny. Sugerujemy wykorzystanie tego algorytmu w celu ostatecznego zakończenia diagnostyki zarówno PWS, jak i AS. Twierdzimy, że diagnoza jest niekompletna bez znajomości specyficznego podtypu genetycznego pacjenta, co pozwala na prowadzenie poradnictwa, przewidywania, postępowania i prawdopodobnych opcji terapeutycznych. Określenie klasy molekularnej jest ważne dla opieki medycznej i leczenia oraz pomocne dla klinicysty zaangażowanego w poradnictwo genetyczne dla członków rodzin z PWS lub ZZSK.
Wkład autorów
MB i JD przyczynili się do sporządzenia manuskryptu, przeglądu literatury, wnieśli swoją wiedzę i zredagowali manuskrypt.
Finansowanie
Uznajemy grant National Institute of Child Health and Human Development o numerze HD02528.
Konflikt interesów
Autorzy oświadczają, że badania były prowadzone przy braku jakichkolwiek komercyjnych lub finansowych powiązań, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Uznajemy Grace Graham za fachowe przygotowanie manuskryptu.
1. Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Postępowanie w zespole Pradera-Williego. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Kliniczne i genetyczne aspekty zespołu Angelmana. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Zespół Pradera-Williego. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Zespół Pradera-Williego: przegląd wyników badań klinicznych, genetycznych i endokrynologicznych. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Pojedyncze geny i syndromiczne przyczyny otyłości: ilustracyjne przykłady. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analysis of the Prader-Willi syndrome imprinting center using droplet digital PCR and next-generation whole-exome sequencing. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Trójka rodzeństwa z zespołem Pradera-Williego spowodowanym przez mikrodelecje centrum imprintingu i przegląd. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Zespół Pradera-Williego i atypowe submikroskopowe delecje 15q11-q13 z lub bez defektów imprintingu. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Zespół Pradera-Williego i zespół Schaafa-Yanga: choroby neurorozwojowe przecinające się w genie MAGEL2. Diseases. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Fenotyp Pradera-Williego spowodowany ojcowskim niedoborem dla klastra HBII-85 C/D box small nucleolar RNA. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-Like phenotype caused by an atypical 15q11.2 microdeletion. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Rodzicielskie pochodzenie delecji chromosomu 15 w zespole Pradera-Williego. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Molekularna klasyfikacja genetyczna w zespole Pradera-Williego: wielomiejscowe badanie kohortowe. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Zespół Pradera-Williego: ustalenia kliniczne i genetyczne. Endocrinologist. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Zespół Pradera-Williego: aktualne rozumienie przyczyny i diagnozy. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Różnice behawioralne wśród osób z zespołem Pradera-Williego i delecją typu I lub typu II oraz matczyną disomią. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Fazy żywieniowe w zespole Pradera-Williego. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct fenotypes distinguish the molecular classes of Zespół Angelmana. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a case-controlled study. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Clinical utility gene card for: Angleman syndrome. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelman syndrome – insights into a rare neurogenetic disorder. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypowy zespół Angelmana spowodowany mozaikowym defektem imprintingu: opisy przypadków i przegląd literatury. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Badanie kliniczne i cytogenetyczne 39 osób z zespołem Pradera-Labharta-Williego. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hipopigmentacja: wspólna cecha zespołu Pradera-Labharta-Williego. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Intellectucal characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. The relationship between compulsive behavior and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Zachowany język ekspresyjny jako fenotypowy wyznacznik mozaikowego zespołu Angelmana. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar
.