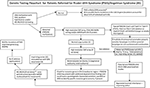
Graphical Abstract. Stroomdiagram voor genetisch onderzoek bij patiënten die zijn doorverwezen voor het Prader-Willi-syndroom (PWS)/Angelman-syndroom (AS). *Verwijder translocaties of omkeringen van Chr15 door routinechromosoomonderzoek; overweeg andere aan obesitas gerelateerde genetische aandoeningen; vereist mogelijk DNA-screening voor het fragiele X-syndroom op FMR1-genherhalingsexpansie of geavanceerde genetische tests met next-generation sequencing (NGS) voor FMR1 of andere kandidaat-genvarianten met behulp van whole-exome sequencing (WES) of whole-genome sequencing (WGS; bv. monogene oorzaken van obesitas). **Kan worden gebruikt om de methyleringsstatus van andere Chr15 ingeprente genen te controleren; ddPCR, droplet digital PCR kan worden gebruikt voor mozaïekscreening.
Inleiding
De chromosoom 15 inprentingsstoornissen omvatten Prader-Willi (PWS) en Angelman (AS) syndromen (1-6) en chromosoom 15q duplicaties. De diagnose van PWS of AS hangt af van de ouder van oorsprong en of de expressie abnormaal beperkt is tot de maternale of de paternale ingeprente genen. Duplicatie 15q wordt veroorzaakt door een extra kopie van de maternale 15q11.2-q13 regio die kan leiden tot epileptische aanvallen, cognitieve en gedragsproblemen waaronder autisme spectrum stoornis (ASS), maar niet tot een PWS of AS fenotype. PWS ontstaat door verlies van maternaal ingeprente en vaderlijk tot expressie komende genen uit de chromosoom 15q11-q13 regio, terwijl AS wordt veroorzaakt door verlies van ingeprente en maternaal tot expressie komende genen in deze regio, waarbij met name het UBE3A gen wordt getroffen. Door de ingeprente aard van de verantwoordelijke genen kunnen zowel genetische als epigenetische fouten de oorzaak zijn.
In 1989 werd bij degenen met zowel PWS als niet-deletie vastgesteld dat de moeder disomie 15 of beide 15s had bij gebruik van polymorfe DNA-merkers uit de proximale 15q11-q13-regio (7). Later in het midden van de jaren negentig werden met de ontwikkeling van fluorescente in situ hybridisatie (FISH) DNA-sondes gebruikt om deleties van de 15q11-q13 regio in zowel PWS als AS te identificeren. Methylatie DNA testen werden in deze periode ontwikkeld en een abnormaal methylatie patroon werd gezien in PWS en AS. Methylatie DNA testen zijn ~99% accuraat in het identificeren van de diagnose PWS, maar zullen niet de individuele moleculaire klasse van PWS identificeren (2). Voor AS identificeren DNA-methyleringstests ~80% van de individuen, maar ook hier wordt geen onderscheid gemaakt tussen de moleculaire klassen of wordt een mutatie in het UBE3A-gen gedetecteerd die AS veroorzaakt.
Microarray-technologie is in het begin tot halverwege de jaren 2000 ontwikkeld en heeft het diagnostische rendement vergroot. Nu omvatten de nieuwe SNP-microarrays meer dan twee miljoen DNA-sondes en zijn ze nuttig voor het opsporen van deletiesubtypes en UPD15-subklassen. Andere technologie zoals druppel digitale PCR (ddPCR) kwantificeert het aantal kopieën met behulp van chromosoom 15 DNA-probes en kan genetische defecten in PWS of AS diagnosticeren (8). Bovendien kunnen SNP-microarrays LOH’s identificeren die gedefinieerd zijn als >8 Mb groot en wanneer aanwezig op chromosoom 15 ondersteunt dit de diagnose van maternale disomie 15 of paternale disomie 15 in aanwezigheid van een abnormaal DNA-methyleringspatroon voor PWS of AS, respectievelijk. Bevestiging van het inprentingsdefect kan niet alleen SNP-microarrays vereisen om kleine microdeleties te identificeren, maar ook ouderlijke DNA-monsters met genotypering om de aanwezigheid van normale (biparentale) overerving van chromosoom 15s vast te stellen die de aanwezigheid van een epimutatie inprentingsdefect bij PWS of AS ondersteunt en daarmee het risico van recidief beïnvloedt. Differentiatie van een IC microdeletie van een niet-deletie epimutatie status is klinisch belangrijk voor families omdat een 50% recidief risico aanwezig is voor extra kinderen als een IC microdeletie wordt gevonden bij de ouder (9).
Er zijn meer dan een dozijn genen en transcripten in de 15q11-q13 regio die een rol lijken te spelen bij de oorzakelijkheid van PWS en/of AS. Genen en transcripten in het gebied tussen de proximale 15q11.2 breukpunten BP1 en het distale 15q13 breukpunt BP3 zijn TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, niet-coderende RNA’s (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2, en HERC2. Ingepente MRKN3, MAGEL2, NDN, NIPAP1, en SNURF-SNRPN genen komen vaderlijk tot expressie en kunnen, wanneer ze verstoord zijn, kenmerken van PWS veroorzaken. Bijvoorbeeld, MAGEL2 genmutaties kunnen neonatale hypotonie, ontwikkelingsachterstand, arthrogryposis, autistische kenmerken, een slechte zuigkracht, en zwaarlijvigheid veroorzaken. Er zijn ook patiënten gemeld met kenmerken van PWS als gevolg van kleine deleties van het niet-coderende SNORD116-transcript (12) en andere soortgelijke deleties in de regio (10, 13).
We hebben ons in dit rapport gericht op AS en PWS omdat beide syndromen worden gedetecteerd via DNA-methyleringstests, waarmee het actieve ouderlijke genallel kan worden bepaald en de definitieve diagnose kan worden gesteld bij personen met PWS en bij de meeste personen met AS (2). Met DNA-methyleringstesten kan echter bij geen van beide syndromen de moleculaire klasse worden vastgesteld. Hoge-resolutie chromosoom analyse werd ontwikkeld en gebruikt in de vroege jaren 1980 en werd een standaard laboratorium genetische-gebaseerde test om te evalueren voor het chromosoom 15q11-q13 deletie geïdentificeerd in de meerderheid van de patiënten met PWS op dat moment (14) en later voor AS. De vaderlijke oorsprong van de 15q11-q13 deletie werd gerapporteerd in 1983 (15) en bleek de novo te zijn, maar de grootte van de 15q11-q13 deletie of het type (typisch vs. atypisch) kon niet worden vastgesteld. Nauwkeurige en vroege diagnose met identificatie van de moleculaire klasse is essentieel, niet alleen om de klinische diagnose te bevestigen, maar ook voor genetische counseling, om de zorg en behandeling te informeren en om de verwachtingen te sturen. Met het oog op lopende klinische trials kan een beter begrip van de moleculaire etiologie van invloed zijn op de mogelijkheden voor patiëntenparticipatie. Verder omvatten op handen zijnde trials antisense oligonucleotiden om de tot zwijgen gebrachte vaderlijke kopie van het chromosoom 15 te reactiveren bij personen met AS.
PWS en AS zijn complexe zeldzame neurologische ontwikkelingsstoornissen als gevolg van fouten in de genomische inprenting. PWS wordt erkend als de meest voorkomende genetische oorzaak van levensbedreigende zwaarlijvigheid, indien ongecontroleerd gelaten (2, 4, 6). Er zijn drie erkende moleculaire klassen van PWS, waaronder een paternale 15q11-q13 deletie van ongeveer 5-6 Mb groot (60% van de gevallen) en maternale disomie 15 (UPD15) waarbij beide chromosomen 15 van de moeder worden geërfd (36%), afkomstig van trisomie 15 met verlies van het paternale chromosoom 15 in de vroege zwangerschap, waardoor twee chromosomen 15 van de moeder worden geërfd (16). De derde categorie is een defect in het inprentingscentrum. Als een microdeletie of epimutatie van het inprentingscentrum (IC), dat de expressiestatus van geselecteerde ingeprente genen op chromosoom 15 controleert, aanwezig is op het vaderlijke allel, dan treedt PWS op. Dit inprentingsdefect wordt bij 4% van de mensen met PWS gezien (8, 16). De meeste gevallen van PWS zijn sporadisch met een geschatte gelijkheid tussen etnische groepen en geslacht. De geschatte prevalentie van PWS is één op 10.000 tot één op 30.000 (2). Het aantal mensen met PWS wereldwijd wordt geschat op ~400.000, waarvan ongeveer 20.000 in de VS (2, 17).
PWS wordt gekenmerkt door infantiele hypotonie, een slechte zuigreflex met voedingsmoeilijkheden, een klein gestalte met kleine handen en voeten, hypogonadisme secundair aan hormoondeficiënties, licht verstandelijke handicap, gedragsproblemen, en hyperfagie vaak met begin tussen 6 en 8 jaar die aanhoudt tot in de volwassenheid en resulteert in obesitas als omgevingscontroles niet op zijn plaats zijn. Tijdens de kinderjaren worden karakteristieke craniofaciale kenmerken gezien, waaronder een smalle bifrontale diameter, strabismus, kleine wipneus met een dunne bovenlip, en naar beneden gedraaide mondhoeken, kleverig speeksel, en glazuurhypoplasie (2, 4, 6, 18). Cognitie is over het algemeen verminderd op basis van de familie achtergrond en gedragsproblemen die beginnen in de kindertijd omvatten zelfverwonding (huid plukken), uitbarstingen, koppigheid, en driftbuien met psychiatrische problemen die optreden tijdens deze periode of later in de adolescentie of jonge volwassenheid (2). Gedragsproblemen omvatten angst, stemmingsstoornissen, psychose en autisme die kunnen correleren met specifieke PWS genetische subtypen of moleculaire klassen (19).
Historisch wordt PWS verdeeld in twee klinische stadia met falen om te gedijen tijdens de kindertijd vertegenwoordigt het eerste klinische stadium en hyperfagie met begin van obesitas vertegenwoordigt het tweede stadium (2). Later zijn voedingsfasen beschreven voor deze aan obesitas gerelateerde genetische aandoening en deze omvatten: Fase 0 met verminderde foetale beweging en groeiachterstand in de baarmoeder, gevolgd door Fase 1 in verband met hypotonie, niet gedijen met moeite met voeden, Fase 2 vanaf ~ 2 jaar wanneer gewichtstoename voor het eerst wordt opgemerkt en Fase 3 wanneer gebrek aan verzadiging gepaard gaat met voedsel zoeken en hyperfagie wat leidt tot obesitas, indien niet extern gecontroleerd. Fase 3 begint rond de leeftijd van 6-8 jaar (20).
Het syndroom van Angelman wordt gekenmerkt door een ontwikkelingsachterstand die vaak pas duidelijk wordt rond de leeftijd van 6 maanden en het daaropvolgende begin van vaak moeilijk onder controle te houden toevallen, tremor, wijdbeens lopen en ataxie met een karakteristieke vrolijke houding (3). Er zijn vier erkende moleculaire mechanismen van AS: de novo maternale deleties van chromosoom 15q11-q13 (70-80%); mutaties van het maternaal overgeërfde UBE3A-gen (10-20%); paternale disomie 15 (3-5%); en imprinting defecten (3-5%) binnen de 15q11-q13 regio die de expressie van het oorzakelijke UBE3A-gen veranderen (21).
Individuen met AS worden vaak niet opgemerkt door medische professionals tot ~6 maanden leeftijd wanneer vertragingen in de ontwikkeling in het bijzonder vertraagde motorische ontwikkeling worden gemeld. Tegen die tijd kunnen ouders de vrolijke houding herkennen, zoals vaak lachen, glimlachen en opgewonden zijn. Een verminderde behoefte aan slaap wordt gemeld in >80% van de individuen met AS (22). Zij ontwikkelen vaak epileptische aanvallen op de leeftijd van 1-3 jaar (23). Epilepsie kan hardnekkig zijn en heeft een karakteristieke verschijning op het EEG beschreven als een verhoogde delta power met een karakteristieke triphasic wave. Individuen met AS worden beschreven als ataxisch in hun bewegingen en lopen (24, 25). Microcefalie kan zich ontwikkelen rond de leeftijd van ~2 jaar. Stereotiep gedrag omvat een liefde voor water en knisperend papier en individuen met AS zijn karakteristiek non-verbaal en gecategoriseerd als ernstig verstandelijk gehandicapt. Het is echter opmerkelijk dat AS-patiënten vaardigheden bezitten die niet goed tot uitdrukking komen in de thans beschikbare objectieve neuropsychologische tests. Ze hebben sterke vaardigheden in het manipuleren van elektronica, maar gedragingen kunnen uitdagend zijn en angst met een korte aandachtsspanne omvatten.
Aangezien patiënten met PWS of AS zich kunnen presenteren met variabele fenotypes afhankelijk van de moleculaire klasse en omdat er voor elk een potentiële behandeling en surveillance aanpak bestaat, is er een logisch stroomschema nodig voor het bestellen van genetische testen door de clinicus die deze patiënten evalueert. De nadruk van ons rapport ligt op het beschrijven van de klinische en genetische bevindingen van deze twee genomische imprinting aandoeningen en het illustreren van genetische testopties die beschikbaar zijn in de klinische setting en de volgorde waarin de verschillende genetische tests het meest productief kunnen worden verkregen.
Laboratorium Genetica Ervaring in Chromosoom 15 Imprinting Stoornissen
Prader-Willi Syndroom
Om als voorbeeld te dienen van het belang van hoge-resolutie SNP microarray testen, werd een groot multisite cohort van 510 deelnemers met genetisch bevestigde PWS gerekruteerd in de VS en gegroepeerd in drie moleculaire klassen. Zij werden verder gekarakteriseerd als 15q11-q13 deletie subtypes, maternale disomie 15 subklassen en imprinting centrum defecten (16). In dit grootste gerapporteerde PWS cohort, bleken 303 personen de 15q11-q13 deletie te hebben (60% van de gevallen), bestaande uit 118 personen (38,9%) met de grotere typische 15q11-q13 Type I deletie waarbij de chromosoom 15q11-q13 breekpunten BP1 en BP3 betrokken waren en 165 personen (54,5%) met de kleinere typische 15q11-q13 deletie.5%) hadden de kleinere typische 15q11-q13 type II deletie waarbij de breekpunten BP2 en BP3 betrokken waren, met 20 personen met een atypische deletie die groter of kleiner is dan de typische 15q11-q13 deletie (6,6%). Bij personen bij wie een deletie van chromosoom 15 is vastgesteld, is het belangrijk te overwegen of een gebalanceerde translocatie aanwezig zou kunnen zijn in de vader van de proband, omdat dit het recidiverisico van PWS in de nakomelingen van de vader verhoogt. Voor maternale disomie 15 hadden 185 individuen (36%) maternale uniparentale disomie 15 (UPD15), waarbij 13 individuen (12,5%) totale isodisomie van het gehele chromosoom 15 hadden als gevolg van fouten in maternale meiose II; 60 (57,7%) vertoonden segmentale isodisomie als gevolg van crossover gebeurtenissen in maternale meiose I en 31 vertoonden heterodisomie (29,8%), terwijl bij 81 individuen geen SNP-microarray analyse en maternale disomie 15 classificatie werd vastgesteld. Wat PWS inprentingsdefecten betreft, werden 22 individuen (4%) gevonden, waarbij 13 (76,5%) een non-deletie epimutatiestatus hadden, vier individuen (23,5%) een microdeletie van het inprentingscentrum hadden, terwijl bij de resterende vijf individuen geen type inprentingsdefect werd vastgesteld. In een verwante studie, werd verdere analyse van imprinting defecten in PWS uitgevoerd door Hartin et al. (8) met behulp van droplet digitale PCR en next-generation whole-exome sequencing in een apart PWS cohort van 15 onverwante patiënten en bij twee individuen of 13% werd een microdeletie van het imprinting centrum defect gevonden. Bij de 60 individuen met segmentale isodisomie 15, gerapporteerd door Butler et al. (16), was de totale gemiddelde grootte van het verlies van heterozygositeit (LOH) 25,1 Mb met een bereik van 5-67,4 Mb en een gemiddelde grootte van 16,4 Mb voor individuele LOHs. Tweeëndertig individuen hadden één LOH segment, 25 individuen hadden twee segmenten en drie individuen hadden drie segmenten. De meest voorkomende LOH-locaties waren de proximale 15q11-q13-regio en de distale 15q26-regio, inclusief de 15q12- en 15q26.1-banden die het vaakst werden geregistreerd.
De aanwezigheid van maternale UPD15 en de bepaling van een specifieke subklasse (segmentale of totale isodisomie) kan van invloed zijn op de diagnose en het toezicht op de medische zorg, omdat er een tweede genetische aandoening aanwezig kan zijn als de moeder drager is van een recessief genallel dat zich in de LOH-regio bevindt en leidt tot twee identieke kopieën. Honderden potentieel ziekteveroorzakende genen worden op chromosoom 15 gevonden en deze ziekten moeten van nabij worden gecontroleerd of bewaakt bij mensen met segmentale of totale isodisomie van chromosoom 15. Een voorgesteld stroomschema voor genetische testen om de verschillende moleculaire klassen voor zowel PWS als AS patiënten te identificeren kan worden gezien in Graphical Abstract.
Angelman Syndrome
Vier erkende moleculaire klassen zijn geïdentificeerd in AS die kunnen worden gecategoriseerd door de impact op de methylering van het chromosoom 15 gebied. Het meest voorkomende subtype is een deletie van het maternale 15q11.2-q13 regio zoals die ook van vaderlijke oorsprong gezien wordt bij PWS en gevonden wordt bij ~70% van de personen met AS (21). Echter, in AS komt de typische klasse II deletie vaker voor. Deze typische kleinere klasse II deletie is meestal ongeveer 5 Mb groot van BP2-BP3 en is aanwezig in 50% van de AS deletie gevallen. Klasse I deleties zijn 5-7 Mb groot en omvatten BP1-BP3 (40% van de deletiegevallen). Atypische deleties kunnen zich uitstrekken van BP1 of BP2-BP4 of verder weg gelegen breekpunten. Bij personen met een deletie op de maternale kopie van chromosoom 15 moet men nagaan of er op de chromosomale microarray aanwijzingen zijn voor verstoringen die wijzen op een mogelijke maternale translocatie. Dit verhoogt het recidiverisico van AS bij toekomstige nakomelingen van de moeder. Uniparentale paternale disomie 15 komt voor bij 5-7% van de personen met AS. Imprinting defecten maken 3-5% uit van individuen met AS en worden veroorzaakt door defecten in het imprinting controle centrum, samengevat door Buiting et al. (26). Bij individuen met een defect in het imprinting controle centrum, slaagt de epigenetische markering in de kiembaan er niet in om op de juiste wijze over te schakelen van een vaderlijk patroon met verzwegen UBE3A expressie naar een maternaal expressiepatroon van het UBE3A gen. In maar liefst 50% van de gerapporteerde gevallen kan een mutatie in het imprinting controle centrum worden geïdentificeerd. Mozaïek gevallen van defecten in het imprinting centrum, waarbij een percentage van de cellen geen expressie van de 15q11.2-q13 regio heeft, zijn gemeld en komen mogelijk vaker voor dan eerder gedacht (27). Het laatste genetische defect in AS heeft geen invloed op de resultaten van DNA methyleringstesten, maar wordt veroorzaakt door een mutatie in het maternaal overervende UBE3A gen. Mutaties in dit gen zijn verantwoordelijk voor 11% van de AS gevallen (28). Een UBE3A mutatie zou maternaal overerfbaar kunnen zijn en daarom is het geïndiceerd om gericht onderzoek te doen bij de moeder van de patiënt om een 50% recidiefrisico bij haar toekomstige nakomelingen uit te sluiten. Als de mutatie wordt geacht te zijn overgeërfd, bevelen wij aan te overwegen de grootvader van moederszijde van de patiënte te testen, aangezien dit gevolgen kan hebben voor de toekomstige kinderen van de tante van moederszijde.
Discussie
De medische behandeling van PWS en AS moet tijdens de zuigelingenleeftijd worden geleid door een multidisciplinair team. Zowel zuigelingen met PWS (vaker voorkomend) als met AS kunnen falen om te gedijen. Een diëtist speelt een belangrijke rol in de zorg in het begin om “failure to thrive” aan te pakken en later in de kindertijd om obesitas te voorkomen met dieet interventie met restrictie en het gebruik van oefenprogramma’s (dit is een punt van zorg dat meer wordt opgemerkt voor PWS, maar nu ook wordt herkend in AS bij sommige individuen). Klinisch genetici, orthopedische specialisten, huisartsen, gespecialiseerde ergotherapeuten (OT), fysiotherapeuten (PT) en spraaktherapeuten (SLP), deskundigen op het gebied van de geestelijke gezondheidszorg, slaapspecialisten, deskundigen op het gebied van de geestelijke gezondheidszorg en endocrinologen zijn nodig om de meervoudige gezondheidsproblemen die zich bij PWS kunnen voordoen, aan te pakken. Een AS team bestaat uit klinisch genetici, neurologen, gespecialiseerde therapeuten voor PT, OT, en SLP diensten, slaapspecialisten, gastro-enterologie, fysische geneeskunde en revalidatie, orthopedie, en geestelijke gezondheidsdeskundigen. Voor PWS zijn de juiste medische zorg, management en begeleiding doelen om gewichtstoename onder controle te houden en geassocieerde comorbide aandoeningen, gedrag en psychiatrische problemen te monitoren en te behandelen. Tekorten aan groeihormonen en andere hormonen die bij deze aandoening veel voorkomen vereisen behandeling. Strikte controle van het dieet met voedselzekerheid en een beheerde routine-omgeving met regelmatige lichaamsbeweging zijn belangrijke strategieën om hyperfagie, obesitas en gerelateerde complicaties onder controle te houden die gedurende het hele leven nodig zijn. AS vereist vroegtijdige interventie met inbegrip van kennis van gespecialiseerde therapeutische interventies zoals augmentatieve en ondersteunende communicatiemiddelen en een versterkend programma van intensieve ontwikkelingsoefeningen en activiteiten voor het bereiken van maximale mogelijkheden (bijv. SPIDER), vroegtijdige behandeling met benzodiazepinen voor aanvallen en dieettherapie zoals het gebruik van een ketogeen dieet. Het maximaliseren van alle aspecten van de zorg, inclusief slaapstoornissen en constipatie, heeft een grote invloed op de controle over aanvallen. Een gespecialiseerd centrum dat bekend is met de fijne kneepjes en unieke aspecten van deze stoornissen kan het resultaat beïnvloeden.
Een vroege diagnose is van vitaal belang voor een vroegtijdige interventie voor zowel PWS als AS. Voor PWS moet een vroege diagnose worden gesteld tijdens de kindertijd om een groeihormoonbehandeling te starten, voedingsproblemen, obesitas, hormoondeficiënties, ontwikkelingsachterstanden en gedragsproblemen te behandelen. De diagnose bij AS zorgt ook voor vroegtijdige therapieën die van invloed zijn op de ontwikkelingsresultaten, en voor profylaxe tegen aanvallen, inclusief voorbereiding met de juiste benzodiazepines. Andere interventies die gunstig kunnen zijn, zijn speciale diëten voor mensen met AS, zoals het ketogeen dieet of lage glycemische index therapie (LGIT). Een vroege diagnose kan ook de kosten van de medische zorg verlagen door het voorkomen van langdurige ziekenhuisopnames in verband met voedingsproblemen bij personen met PWS en epileptische aanvallen bij kinderen met AS.
Het identificeren van de moleculaire klasse van PWS of AS met geavanceerde genetische tests zoals hoge-resolutie SNP-microarrays zal een nauwkeuriger diagnose mogelijk maken, wat leidt tot betere indicatoren voor prognose, en nauwkeuriger genetische counseling van familieleden. SNP-microarrays met hoge resolutie, FISH-analyse, methyleringsspecifieke-multiplex-ligatieprobe-amplificatie (MS-MLPA), en/of genotypering van chromosoom 15 zijn alle nuttig voor het vaststellen van 15q11-q13 deleties. Hoge-resolutie SNP-microarrays kunnen de deletiesubtypes (typisch en atypisch) in zowel PWS als AS, en UPD15-subklassen (segmentale isodisomie, en totale isodisomie) identificeren. Het heterodisomie subtype van UPD en IC defecten (microdeletie en epimutatie) bij zowel PWS als AS kan extra diagnostisch werk vereisen, zoals geïllustreerd in Graphical Abstract. Het subtype of de klassen hebben invloed op de diagnose, het potentiële recidief risico voor familieleden, de prognose en de controle op andere genetische aandoeningen en hoog-risico kenmerken gerelateerd aan de moleculaire klasse. Autistische kenmerken en psychose komen bijvoorbeeld vaker voor bij mensen met PWS en maternale disomie 15 en kunnen verband houden met de specifieke UPD15-subklassen. Degenen met de grotere klasse I deleties in AS hebben meer kans op het ontwikkelen van moeilijk te behandelen epileptische aanvallen en microcefalie.
Een stroomdiagram voor genetisch testen met de beschikbare testopties, inclusief de opties die in het verleden voor zowel PWS als AS werden gebruikt, is opgenomen in het grafische abstract. Het testen op PWS of AS begint vaak met DNA methylering en indien abnormaal wordt dan verder gegaan met andere genetische testmethoden waaronder hoge-resolutie SNP microarrays of MS-MLPA assays gebaseerd op de beschikbaarheid voor de clinici en families in hun klinische setting. Bij voorkeur wordt een hoge-resolutie SNP-array besteld die in de westerse medische zorg gemakkelijk en commercieel verkrijgbaar is. Next-generation sequencing (NGS) van het exoom (of het volledige genoom) is ook beschikbaar voor clinici, maar droplet digital PCR (ddPCR) is momenteel gebaseerd op onderzoek (14). SNP arrays kunnen specifieke moleculaire klassen identificeren bij de meerderheid van de patiënten die zich presenteren met kenmerken van PWS (ongeveer 85% van de gevallen) of AS (ongeveer 80%), terwijl de resterende patiënten aanvullende testen nodig zullen hebben zoals beschreven in Graphical Abstract. Specifieke geavanceerde genetische testen (bijv. ddPCR) kunnen voldoende gevoelig zijn om mozaïcisme te kwantificeren en kunnen een diagnose identificeren in een grote subset van individuen met mildere klinische kenmerken van PWS en AS, maar meer onderzoek is nodig.
Er werden vroege klinische verschillen gevonden bij het vergelijken van die met PWS of AS die de deletie versus niet-deletie status hadden (29) inclusief hypopigmentatie bij die met PWS en AS die de 15q11-q13 deletie hadden (30). Later werden hogere verbale IQ scores (31) of psychose (32) gerapporteerd bij diegenen met maternale UPD15 vergeleken met deletie bij personen met PWS. Verder rapporteerden Butler et al. (19) lagere adaptieve scores en meer obsessief-compulsief gedrag bij PWS individuen met de 15q11-q13 Type I deletie in vergelijking met UPD15. Zarcone et al. (33) rapporteerden dat personen met PWS en de 15q11-q13 Type I deletie meer dwanghandelingen hadden met persoonlijke netheid en dwangmatig gedrag dat moeilijk te onderbreken was en meer interfereerde met sociale activiteiten dan die met Type II deleties of UPD15. In een fenotype-genotype correlatie studie bij het Angelman syndroom, rapporteerden Moncla et al. (34) verhoogde aanvalsactiviteit bij degenen met de grotere klasse I deletie in vergelijking met niet-deletie. Microcefalie, ataxie, hypotonie, en voedingsproblemen zijn ook meer waarschijnlijk in het deletie subtype (3). Zij kunnen ernstigere taalstoornissen hebben, in het bijzonder receptieve taal en autistische trekken (21, 35). Personen met AS met UPD van vaderskant kunnen een verbeterde receptieve taal hebben, verbeterde motoriek, en een verminderde prevalentie van aanvallen. Mozaïek individuen kunnen ook een milder fenotype hebben, inclusief verbeterde taalvaardigheden, adaptief functioneren, en minder aanvallen (36).
Next-generation exome of whole-genome sequencing kan ook een plaats hebben in genetische evaluaties van PWS of AS, vooral bij die individuen die zich presenteren met ongewone bevindingen of vertraagde diagnose (bijv. UPD15 segmentale of totale isodisomie) en in gevallen waarin ouderlijk DNA niet beschikbaar is (8). Om het gebruik en het type van genetische testen voor PWS en AS aan te pakken, werd een nieuw stroomdiagram voor genetische testen ontwikkeld voor de clinicus zoals beschreven en geïllustreerd in Graphical Abstract. Dit stroomschema kan helpen bij het bestellen van genetische testen op basis van klinische presentatie om de juiste diagnose, management en behandeling te bepalen en om de meest accurate genetische counseling informatie voor andere familieleden te leveren. Wij stellen het gebruik van dit algoritme voor om het diagnostisch werk voor zowel PWS als AS definitief te voltooien. Wij stellen dat de diagnose onvolledig is zonder kennis van het specifieke genetische subtype van de patiënt als leidraad voor counseling, anticiperende begeleiding, management en waarschijnlijke therapeutische opties. De bepaling van de moleculaire klasse is belangrijk voor de medische zorg en behandeling en nuttig voor de clinicus die betrokken is bij de genetische counseling van familieleden voor PWS of AS.
Bijdragen van auteurs
MB en JD hebben bijgedragen aan het opstellen van het manuscript, het beoordelen van de literatuur, hebben hun expertise ingebracht en hebben het manuscript geredigeerd.
Funding
Wij erkennen het National Institute of Child Health and Human Development subsidienummer HD02528.
Conflict of Interest
De auteurs verklaren dat het onderzoek werd uitgevoerd in afwezigheid van enige commerciële of financiële relaties die zouden kunnen worden opgevat als een potentieel belangenconflict.
Acknowledgments
Wij erkennen Grace Graham voor de deskundige voorbereiding van het manuscript.
1. Bittel DC, Butler MG. Prader-Willi syndroom: klinische genetica, cytogenetica en moleculaire biologie. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Management van het Prader-Willi-syndroom. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Klinische en genetische aspecten van het Angelman syndroom. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willi syndroom. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Single gene and syndromic causes of obesity: illustrative examples. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analyse van het Prader-Willi syndroom imprinting centrum met behulp van droplet digitale PCR en volgende generatie whole-exome sequencing. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Three siblings with Prader-Willi syndrome caused by imprinting center microdeletions and review. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Prader-Willi syndroom en atypische submicroscopische 15q11-q13 deleties met of zonder imprinting defecten. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fontein MD, Schaaf CP. Prader-Willi-syndroom en Schaaf-Yang-syndroom: neurologische ontwikkelingsziekten die elkaar kruisen bij het MAGEL2-gen. Diseases. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi fenotype veroorzaakt door paternal deficiëntie voor de HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-Like phenotype veroorzaakt door een atypische 15q11.2 microdeletie. Genen (Bazel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deleties van chromosoom 15 als een oorzaak van het Prader-Willi syndroom. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Moleculair genetische classificatie in Prader-Willi syndroom: een multisite cohort studie. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Prader-Willi syndroom: klinische en genetische bevindingen. Endocrinologist. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Onderscheidende fenotypes onderscheiden de moleculaire klassen van het Angelman syndroom. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a case-controlled study. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Klinische bruikbaarheid genenkaart voor: Angleman syndroom. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndroom. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelman syndroom – inzichten in een zeldzame neurogenetische aandoening. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypical Angelman syndrome due to a mosaic imprinting defect: case reports and review of the literature. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Clinical and cytogenic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hypopigmentatie: een gemeenschappelijk kenmerk van Prader-Labhart-Willi syndroom. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Intellectucal characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. The relationship between compulsive behavior and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preserved expressive language as a phenotypic determinant of mosaic Angelman syndrome. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar