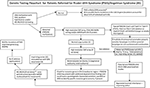
Graphical Abstract. Genetikai vizsgálati folyamatábra Prader-Willi-szindróma (PWS)/Angelman-szindróma (AS) miatt beutalt betegeknél. *Rutin kromoszóma-vizsgálatokkal kizárni a Chr15 transzlokációkat vagy inverziókat; mérlegelni más, elhízással összefüggő genetikai rendellenességeket; szükség lehet törékeny X-szindrómás DNS-szűrésre az FMR1 génismétlődés kiterjesztésére, vagy fejlett genetikai vizsgálatra az FMR1 vagy más jelölt génváltozatokra következő generációs szekvenálással (NGS) a teljes exom szekvenálás (WES) vagy a teljes genom szekvenálás (WGS; pl. az elhízás monogén okai) segítségével. **Csak más Chr15 imprintált gének metilációs státuszának ellenőrzésére használható; ddPCR, droplet digital PCR használható mozaikizmus szűrésére.
Bevezetés
A 15. kromoszóma imprinting rendellenességei közé tartoznak a Prader-Willi (PWS) és az Angelman (AS) szindrómák (1-6) és a 15q kromoszóma duplikációk. A PWS vagy AS diagnózisa a származási szülőtől függ, és attól, hogy az expresszió rendellenesen az anyai vagy az apai imprintelt génekre korlátozódik. A 15q duplikációt az anyai eredetű 15q11.2-q13 régió egy további példánya okozza, ami görcsrohamokhoz, kognitív és viselkedési problémákhoz vezethet, beleértve az autizmus spektrumzavart (ASD), de nem PWS vagy AS fenotípushoz. A PWS a 15q11-q13 kromoszóma 15q11-q13 régiójában található anyai imprintált és apai expressziójú gének elvesztéséből ered, míg az AS-t az ebben a régióban található imprintált és anyai expressziójú gének elvesztése okozza, amely különösen az UBE3A gént érinti. A felelős gének imprintelt jellege miatt mind genetikai, mind epigenetikai hibák okozói lehetnek.
1989-ben a PWS-ben és a nem deléciós státuszban szenvedőknél az anyai 15-ös diszómia vagy mindkét 15-ös az anyától származott, amikor a proximális 15q11-q13 régióból származó polimorf DNS-markereket használtak (7). Később, az 1990-es évek közepén a fluoreszcens in situ hibridizáció (FISH) DNS-szondák kifejlesztésével a 15q11-q13 régió delécióit azonosították mind PWS-ben, mind AS-ben. Ebben az időszakban fejlesztették ki a metilációs DNS-vizsgálatokat, és rendellenes metilációs mintázatot észleltek PWS-ben és AS-ben. A metilációs DNS-vizsgálat ~99%-os pontossággal azonosítja a PWS diagnózisát, de nem azonosítja az egyes PWS molekuláris osztályokat (2). AS esetében a metilációs DNS-vizsgálat az egyének ~80%-át azonosítja, de ismét nem tesz különbséget a molekuláris osztályok között, és nem mutat ki AS-t okozó mutációt az UBE3A génben.
A 2000-es évek elején-közepén kifejlesztett microarray-technológia továbbfejlesztette a diagnosztikai hozamot. Ma már az új SNP-mikrotáblák több mint kétmillió DNS-szondát tartalmaznak, és hasznosak a deléciós altípusok és az UPD15 alosztályok kimutatásában. Más technológiák, mint például a cseppnyi digitális PCR (ddPCR) a 15-ös kromoszóma DNS-szondák segítségével számszerűsíti a kópiaszámot, és képes diagnosztizálni a PWS vagy AS genetikai hibáit (8). Továbbá, az SNP-mikrotáblák képesek azonosítani a >8 Mb méretben meghatározott LOH-okat, és amennyiben a 15-ös kromoszómán jelen vannak, támogatják az anyai 15-ös diszómia vagy az apai 15-ös diszómia diagnózisát a PWS, illetve az AS abnormális DNS-metilációs mintázatának jelenlétében. Az imprinting-defektus megerősítéséhez nemcsak SNP-mikroelemzésekre lehet szükség a kis mikrodeléciók azonosításához, hanem szülői DNS-mintákra is genotipizálással a 15-ös kromoszóma normál (biparentális) öröklődésének azonosításához, ami alátámasztja az epimutációs imprinting-defektus jelenlétét PWS-ben vagy AS-ben, ami hatással van a kiújulás kockázatára. Az IC mikrodeléció és a nem-deléciós epimutációs státusz megkülönböztetése klinikailag fontos a családok számára, mivel további gyermekek esetében 50%-os kiújulási kockázat áll fenn, ha a szülőnél IC mikrodeléciót találnak (9).
A 15q11-q13 régióban több mint egy tucat gén és transzkriptum található, amelyek a jelek szerint szerepet játszanak a PWS és/vagy AS okozásában. A proximális 15q11.2 töréspont BP1 és a distális 15q13 töréspont BP3 közötti területen található gének és transzkriptek a következők: TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, nem kódoló RNS-ek (SNORD), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 és HERC2. Az imprintelt MRKN3, MAGEL2, NDN, NIPAP1 és SNURF-SNRPN gének paternálisan expresszálódnak, és ha megzavarják őket, a PWS jellemzőit okozhatják. Például a MAGEL2 génmutációk újszülöttkori hipotóniát, fejlődési késést, arthrogryposist, autisztikus vonásokat, rossz szopást és elhízást okozhatnak . A nem kódoló SNORD116 transzkriptum kis deléciói (12) és más hasonló deléciók következtében a PWS jellemzőit mutató betegekről is beszámoltak (10, 13).
Ebben a jelentésben az AS-re és a PWS-re összpontosítottunk, mivel mindkét szindróma kimutatható DNS-metilációs vizsgálattal, amely lehetővé teszi az aktív szülői génallél meghatározását és a végleges diagnózist a PWS-ben és a legtöbb AS-ben szenvedő egyénnél (2). A DNS-metilációs vizsgálat azonban egyik szindróma esetében sem azonosítja a molekuláris osztályt. A nagy felbontású kromoszómaanalízist az 1980-as évek elején fejlesztették ki és alkalmazták, és az akkoriban a PWS-ben szenvedő betegek többségénél azonosított 15q11-q13 kromoszóma-deléció értékelésére szolgáló standard laboratóriumi genetikai alapú vizsgálat lett (14), később pedig az AS esetében. A 15q11-q13 deléció apai eredetéről 1983-ban számoltak be (15), és de novo-nak találták, de a 15q11-q13 deléció méretét vagy típusát (tipikus vs. atipikus) nem tudták meghatározni. A pontos és korai diagnózis a molekuláris osztály azonosításával nemcsak a klinikai diagnózis megerősítéséhez, hanem a genetikai tanácsadáshoz, a gondozás és a kezelés tájékoztatásához és az elvárások irányításához is elengedhetetlen. A folyamatban lévő klinikai vizsgálatok szándékával a molekuláris etiológia jobb megértése hatással lehet a betegek részvételének lehetőségeire. Továbbá a küszöbön álló kísérletek között szerepelnek antisense oligonukleotidok, amelyek a 15-ös kromoszóma elnémított apai kópiáját reaktiválják AS-ben szenvedő egyéneknél.
A PWS és az AS a genomi imprinting hibái miatt kialakuló komplex ritka idegfejlődési rendellenességek. A PWS-t az életet veszélyeztető elhízás leggyakoribb genetikai okaként ismerik el, ha nem kontrollálják (2, 4, 6). Három elismert PWS molekuláris osztály van, beleértve az apai 15q11-q13 deléciót, amely körülbelül 5-6 Mb méretű (az esetek 60%-ában) és az anyai 15-ös diszómia (UPD15), amelyben mindkét 15-ös kromoszóma az anyától öröklődik (36%), amely a 15-ös kromoszóma triszómiájából ered, az apai 15-ös kromoszóma korai terhességben történő elvesztésével, ami az anyától származó két 15-ös kromoszómához vezet (16). A harmadik osztály az imprinting-központ hibája. Ha a 15-ös kromoszómán lévő kiválasztott imprinting-gének kifejeződési állapotát szabályozó imprinting-központ (IC) mikrodeléciója vagy epimutációja van jelen az apai allélon, akkor PWS lép fel. Ez az imprinting-hiba a PWS-ben szenvedő egyének 4%-ánál fordul elő (8, 16). A PWS legtöbb esete sporadikus, az etnikai csoportok és a nemek között megközelítőleg egyenlő arányban fordul elő. A PWS becsült előfordulási gyakorisága egy a 10 000-ből egy a 30 000-ből (2). A PWS-ben szenvedő egyének számát világszerte ~400 000-re becsülik, ebből körülbelül 20 000 egyén él az USA-ban (2, 17).
A PWS-t csecsemőkori hipotónia, gyenge szopási reflex és táplálkozási nehézségek, alacsony termet, kis kezek és lábak, hormonhiány miatt másodlagos hipogonadizmus, enyhe értelmi fogyatékosság, viselkedési problémák és hiperfágia jellemzi, amely gyakran 6 és 8 éves kor között kezdődik, és felnőttkorban is fennáll, és elhízáshoz vezet, ha a környezeti kontroll nem működik. Csecsemőkorban jellegzetes craniofaciális jegyek láthatók, beleértve a keskeny bifrontális átmérőt, a kancsalságot, a kis felfelé forduló orrot vékony felső ajakkal és lefelé forduló szájsarkakkal, ragadós nyállal és zománchipopláziával (2, 4, 6, 18). A kogníció a családi háttér alapján általában csökkent, és a gyermekkorban kezdődő viselkedési problémák közé tartozik az önkárosítás (bőrpiszkálás), a kitörések, a makacsság és a dühkitörések, a pszichiátriai problémák pedig ebben az időszakban vagy később, serdülőkorban vagy fiatal felnőttkorban jelentkeznek (2). A viselkedési problémák közé tartozik a szorongás, a hangulatzavarok, a pszichózis és az autizmus, amelyek korrelálhatnak a PWS specifikus genetikai altípusaival vagy molekuláris osztályaival (19).
A PWS-t történelmileg két klinikai szakaszra osztják: az első klinikai szakaszt a csecsemőkori sikertelenség, a második szakaszt pedig a hiperfágia és az elhízás kialakulása jelenti (2). Később táplálkozási fázisokat írtak le erre az elhízással összefüggő genetikai rendellenességre vonatkozóan, amelyek a következők: A 0. fázis csökkent magzati mozgással és méhen belüli növekedési elmaradással jár, ezt követi az 1. fázis, amely hipotóniával és táplálkozási nehézségekkel járó sikertelenséggel jár, a 2. fázis ~2 éves korban kezdődik, amikor először figyelhető meg súlygyarapodás, és a 3. fázis, amikor a jóllakottság hiánya ételkereséssel és hiperfágiával jár együtt, ami elhízáshoz vezet, ha nincs külsőleg kontrollálva. A 3. fázis körülbelül 6-8 éves korban kezdődik (20).
Az Angelman-szindrómára jellemző a fejlődési késés, amely gyakran csak 6 hónapos korban jelentkezik, majd ezt követően gyakran nehezen kontrollálható rohamok, remegés, széles alapú járás és ataxia jelentkezik, jellegzetes vidám viselkedés mellett (3). Az AS négy elismert molekuláris mechanizmusa ismert: a 15q11-q13 kromoszóma de novo anyai deléciója (70-80%); az anyai öröklődő UBE3A gén mutációi (10-20%); az apai 15 diszómia (3-5%); és a 15q11-q13 régióban lévő imprinting-hibák (3-5%), amelyek megváltoztatják az okozó UBE3A gén expresszióját (21).
Az AS-ben szenvedő egyéneket az egészségügyi szakemberek gyakran csak ~6 hónapos korukban veszik észre, amikor a fejlődésben, különösen a késleltetett motoros fejlődésben mutatkozó késésekről számolnak be. Ekkorra a szülők felismerhetik a vidám viselkedést, amely magában foglalja a gyakori nevetést, mosolygást és izgatottságot. Az AS-ben szenvedő egyének >80%-ánál csökkent alvásigényről számolnak be (22). Náluk 1-3 éves korban gyakran alakulnak ki rohamok (23). Az epilepszia lehet nehezen kezelhető, és az EEG-n jellegzetes megjelenést mutat, amelyet megnövekedett delta teljesítményként írnak le, jellegzetes trifázisos hullámmal. Az AS-ben szenvedő egyének mozgása és járása ataxiás (24, 25). A mikrokefália ~2 éves korig kialakulhat. A sztereotip viselkedésformák közé tartozik a víz és a gyűrött papír szeretete, és az AS-ben szenvedő egyének jellemzően nonverbálisak és súlyosan értelmi fogyatékosnak minősülnek. Figyelemre méltó azonban, hogy az AS-ben szenvedő egyének olyan képességekkel rendelkeznek, amelyeket a jelenleg rendelkezésre álló objektív neuropszichológiai tesztek nem tudnak jól leképezni. Erős képességekkel rendelkeznek az elektronika manipulálásában, de a viselkedésük kihívást jelenthet, és magában foglalja a rövid figyelmi idővel járó szorongást.
Mivel a PWS-ben vagy AS-ben szenvedő betegek a molekuláris osztálytól függően változó fenotípussal jelentkezhetnek, és mivel mindegyik esetében léteznek lehetséges kezelési és felügyeleti megközelítések, logikus folyamatábrára van szükség ahhoz, hogy az ezeket a betegeket értékelő klinikusok genetikai vizsgálatokat rendeljenek. Beszámolónk középpontjában e két genomiális imprinting rendellenesség klinikai és genetikai leleteinek leírása áll, valamint a klinikai környezetben rendelkezésre álló genetikai vizsgálati lehetőségek és a különböző genetikai vizsgálatok legproduktívabb sorrendjének bemutatása.
Laborgenetikai tapasztalatok a 15. kromoszóma imprinting-rendellenességekről
Prader-Willi-szindróma
A nagy felbontású SNP-mikroarray-vizsgálatok fontosságának példájaként egy nagy, több helyszínen végzett, 510, genetikailag igazolt PWS-ben szenvedő résztvevőből álló kohorszot toboroztak az USA-ban, és három molekuláris osztályba sorolták őket. Ezeket tovább jellemezték 15q11-q13 deléciós altípusok, anyai diszómia 15 alosztályok és imprinting-központ-hibák szerint (16). Ebben a legnagyobb bejelentett PWS-kohortban 303 egyénnél találtak 15q11-q13-deléciót (az esetek 60%-a), amelyből 118 egyén (38,9%) a 15q11-q13 kromoszóma BP1 és BP3 töréspontjait érintő, nagyobb, tipikus 15q11-q13 I. típusú delécióval, 165 egyén (54.5%) a kisebb, tipikus 15q11-q13 II. típusú delécióval rendelkezett, amely a BP2 és BP3 töréspontokat érinti, és 20 egyénnél (6,6%) a tipikus 15q11-q13 deléciónál nagyobb vagy kisebb atipikus deléciót találtak. A 15-ös kromoszóma deléciójával azonosított személyeknél fontos megfontolni, hogy lehet-e kiegyensúlyozott transzlokáció a próbandum apjában, mivel ez növeli a PWS kiújulásának kockázatát az apa utódaiban. A 15-ös kromoszóma anyai diszómia esetében 185 személynél (36%) volt anyai uniparentális 15-ös diszómia (UPD15), 13 személynél (12,5%) a teljes 15-ös kromoszóma teljes izodiszómiája az anyai meiózis II-ben bekövetkezett hibák miatt; 60 személynél (57,7%) az anyai meiózis I-ben bekövetkezett kereszteződésből származó szegmentális izodiszómia, 31 személynél pedig heterodiszómia (29,8%), míg 81 személynél nem volt SNP microarray elemzés és anyai diszómia 15 besorolás. A PWS imprinting-hibák tekintetében 22 egyednél (4%) találtak imprinting-hibát, amelyek közül 13 (76,5%) nem deléciós epimutációs státusszal rendelkezett, négy egyednél (23,5%) az imprinting-központ mikrodeléciója volt, míg a fennmaradó öt egyednél nem állapították meg az imprinting-hiba típusát. Egy kapcsolódó tanulmányban Hartin és munkatársai (8) a PWS imprinting-hibák további elemzését végezték el cseppnyi digitális PCR és újgenerációs teljes exom-szekvenálás segítségével egy külön PWS-kohorszban, 15 nem rokon betegben, és két egyénnél vagy 13%-nál találtak imprinting-központ mikrodeléciós hibát. A Butler és munkatársai (16) által közölt 60, szegmentális 15-ös izodiszómiával rendelkező egyénnél a heterozigóziavesztés (LOH) teljes átlagos mérete 25,1 Mb volt, 5-67,4 Mb-os tartományban, és az egyes LOH-ok átlagos mérete 16,4 Mb volt. Harminckét egyednél egy LOH szegmens, 25 egyednél két szegmens és három egyednél három szegmens volt. A leggyakoribb LOH-helyek a proximális 15q11-q13 régió és a disztális 15q26 régió voltak, beleértve a 15q12 és 15q26.1 sávokat, mint leggyakrabban rögzítetteket.
Az anyai UPD15 jelenléte és a specifikus alosztály (szegmentális vagy teljes izodiszómia) meghatározása hatással lehet a diagnózisra és az orvosi ellátás felügyeletére, mivel egy második genetikai állapot is jelen lehet, ha az anya a LOH régióban található recesszív génallél hordozója, ami két azonos másolathoz vezet. A 15-ös kromoszómán több száz potenciálisan betegséget okozó gén található, és ezeket a betegségeket a 15-ös kromoszóma szegmentális vagy teljes izodiszómiájával rendelkezőknél szorosan ellenőrizni vagy nyomon követni kell. Egy javasolt genetikai vizsgálati folyamatábra a különböző molekuláris osztályok azonosítására mind a PWS, mind az AS betegek esetében a grafikus kivonatban látható.
Angelman-szindróma
Az AS-ben négy elismert molekuláris osztályt azonosítottak, amelyeket a 15-ös kromoszóma régió metilációjára gyakorolt hatás alapján lehet kategorizálni. A leggyakoribb altípus az anyai 15q11 deléciója.2-q13 régióban, ahogy az apai eredetű PWS-ben is megfigyelhető, és az AS-ben szenvedő egyének ~70%-ánál megtalálható (21). AS-ben azonban a tipikus II. osztályú deléció gyakoribb. Ez a tipikus kisebb II. osztályú deléció leggyakrabban megközelíti az 5 Mb-ot a BP2-BP3 között, és a deléciós AS-esetek 50%-ában fordul elő. Az I. osztályú deléciók 5-7 Mb méretűek és a BP1-BP3-at foglalják magukban (a deléciós esetek 40%-a). Az atipikus deléciók kiterjedhetnek a BP1 vagy BP2-BP4 vagy távolabbi töréspontokra is. A 15. kromoszóma anyai kópiáján deléciót mutató egyéneknél figyelembe kell venni, hogy a kromoszóma-mikroarray-n vannak-e olyan zavarokat mutató jelek, amelyek arra utalnak, hogy anyai transzlokációról lehet szó. Ez növeli az AS kiújulási kockázatát a jövőbeli anyai utódokban. A 15-ös apai diszómia az AS-ben szenvedő egyének 5-7%-át teszi ki. Az AS-ben szenvedő egyének 3-5%-át teszik ki az imprinting-hibák, amelyeket a Buiting és munkatársai által összefoglalt imprinting-ellenőrző központ hibái okoznak (26). Az imprint kontrollközpont hibájával rendelkező egyéneknél az epigenetikus jelölés a csíravonalban nem tud megfelelően átváltani az apai mintázatról az elnémított UBE3A expresszióra, hogy lehetővé tegye az UBE3A gén anyai mintázatú expresszióját. A bejelentett esetek akár 50%-ában azonosítható mutáció az imprinting kontrollközpontban. Az imprinting-központ hibáinak mozaikos eseteit, amelyekben a sejtek egy százalékában hiányzik a 15q11.2-q13 régió expressziója, jelentették, és ez gyakoribb lehet, mint korábban gondolták (27). Az AS végső genetikai hibája nem befolyásolja a DNS-metilációs vizsgálati eredményeket, hanem az anyai öröklésű UBE3A gén mutációja okozza. E gén mutációi az AS-esetek 11%-át teszik ki (28). Az UBE3A mutáció anyai úton öröklődhet, ezért a beteg édesanyjánál célzott vizsgálatot kell végezni, hogy kizárható legyen az 50%-os kiújulási kockázat a későbbi utódoknál. Ha a mutáció öröklődőnek bizonyul, javasoljuk a beteg anyai nagyapjának vizsgálatának megfontolását, mivel ez hatással lehet az anyai nagynéni jövőbeli gyermekeire.
Diszkusszió
A PWS és az AS orvosi kezelését csecsemőkorban egy multidiszciplináris csapatnak kell irányítania. Mind a PWS-ben (gyakrabban), mind az AS-ben szenvedő csecsemőknél előfordulhat sikertelenség. A dietetikus fontos szerepet játszik a gondozásban, először a sikertelenség kezelésében, majd később gyermekkorban az elhízás elkerülése érdekében, diétás beavatkozással, korlátozással és mozgásprogramok alkalmazásával (ami a PWS esetében gyakrabban észlelt, de mostanában az AS esetében is felismert probléma néhány egyénnél). Klinikai genetikusokra, ortopéd szakorvosokra, alapellátó orvosokra, speciális foglalkozás- (OT), fizikoterapeutákra (PT) és logopédusokra (SLP), mentálhigiénés szakemberekre, alvásszakértőkre, mentálhigiénés szakemberekre és endokrinológusokra van szükség a PWS-ben előforduló számos egészségügyi probléma kezeléséhez. Egy AS-csoportban klinikai genetikusok, neurológusok, speciális terapeuták a PT, OT és SLP szolgáltatásokhoz, alvásszakértők, gasztroenterológusok, fizikoterapeuták és rehabilitációs szakemberek, ortopédusok és mentálhigiénés szakértők vesznek részt. A PWS esetében a megfelelő orvosi ellátás, kezelés és tanácsadás célja a súlygyarapodás ellenőrzése, valamint a társuló társbetegségek, viselkedési és pszichiátriai problémák figyelemmel kísérése és kezelése. Az ebben a rendellenességben gyakori növekedési és egyéb hormonhiányok kezelést igényelnek. A táplálkozás szigorú ellenőrzése az élelmiszerbiztonsággal és az irányított rutinszerű környezet rendszeres testmozgással fontos stratégiák a hiperfágia, az elhízás és a kapcsolódó szövődmények ellenőrzésére, melyekre egész életen át szükség van. Az AS korai beavatkozást igényel, beleértve az olyan speciális terápiás beavatkozások ismeretét, mint az augmentatív és segítő kommunikációs eszközök és a maximális lehetőségek elérését célzó intenzív fejlesztő gyakorlatokból és tevékenységekből álló erősítő program (pl. SPIDER), a rohamok kezelésére szolgáló benzodiazepinekkel való korai kezelés és diétás terápia, mint például a ketogén étrend alkalmazása. Az ellátás minden szempontjának maximalizálása, beleértve az alvászavarokat és a székrekedést is, nagyban befolyásolja a rohamok kontrollját. Az e rendellenességek bonyolultságát és egyedi aspektusait ismerő, erre szakosodott központ befolyásolhatja az eredményt.
A korai diagnózis létfontosságú a korai beavatkozás biztosításához mind a PWS, mind az AS esetében. A PWS esetében a korai diagnózist csecsemőkorban kell felállítani a növekedési hormonkezelés megkezdése, a táplálkozási problémák, az elhízás, a hormonhiány, a fejlődési késések és a viselkedési problémák kezelése érdekében. Az AS diagnózisa biztosítja a korai terápiákat is, amelyek hatással vannak a fejlődési eredményekre, valamint a rohamprofilaxist, beleértve a megfelelő benzodiazepinekkel való felkészülést. Egyéb, előnyösnek bizonyuló beavatkozások közé tartoznak az AS-ben szenvedő egyének számára speciális diéták, mint például a ketogén diéta vagy az alacsony glikémiás indexű terápia (LGIT). A korai diagnózis az orvosi ellátás költségeit is csökkentheti azáltal, hogy a PWS-ben szenvedő egyéneknél megelőzi a táplálkozási problémákkal kapcsolatos hosszabb kórházi kezeléseket, az AS-ben szenvedő gyermekeknél pedig a rohamokat.
A PWS vagy AS molekuláris osztályának azonosítása fejlett genetikai vizsgálatokkal, például nagyfelbontású SNP-mikroelemzésekkel pontosabb diagnózist tesz lehetővé, ami a prognózis jobb mutatóihoz és a családtagok pontosabb genetikai tanácsadásához vezet. A 15q11-q13 deléciók meghatározásában hasznosak a nagy felbontású SNP-mikrotáblák, a FISH-analízis, a metiláció-specifikus-multiplex ligációs szonda amplifikáció (MS-MLPA) és/vagy a 15. kromoszóma genotipizálása. A nagy felbontású SNP-mikroszűrőkkel azonosíthatók a deléciós altípusok (tipikus és atipikus) mind a PWS-ben, mind az AS-ben, valamint az UPD15 alosztályai (szegmentális izodiszómia és teljes izodiszómia). Az UPD heterodiszómia altípusa és az IC-hibák (mikrodeléció és epimutáció) mind a PWS-ben, mind az AS-ben további diagnosztikai munkát igényelhetnek, amint azt a grafikus összefoglaló szemlélteti. Az altípus vagy osztályok befolyásolják a diagnózist, a családtagok lehetséges kiújulási kockázatát, a prognózist és az egyéb genetikai állapotok és a molekuláris osztályhoz kapcsolódó magas kockázatú jellemzők nyomon követését. Például az autisztikus vonások és a pszichózis gyakrabban fordulnak elő PWS-ben és anyai diszómia 15-ben szenvedőknél, és ez összefügghet az UPD15 specifikus alosztályaival. Az AS-ben a nagyobb I. osztályú deléciókkal rendelkezőknél nagyobb valószínűséggel alakulnak ki nehezen kezelhető rohamok és mikrokefália.
A grafikus összefoglaló tartalmazza a genetikai vizsgálatok folyamatábráját, amely tartalmazza a rendelkezésre álló vizsgálati lehetőségeket, beleértve a PWS és az AS esetében történetileg használtakat is. A PWS vagy AS vizsgálata gyakran a DNS-metilációval kezdődik, és ha abnormális, akkor a klinikusok és a családok klinikai környezetben való elérhetősége alapján más genetikai vizsgálati módszereket alkalmaznak, beleértve a nagy felbontású SNP-mikroelemeket vagy az MS-MLPA-teszteket. Lehetőleg olyan nagyfelbontású SNP-mintázatot rendelnének, amely a nyugatiasodott orvosi ellátásban könnyen és kereskedelmi forgalomban elérhető. Az exom (vagy a teljes genom) következő generációs szekvenálása (NGS) szintén elérhető a klinikusok számára, de a cseppnyi digitális PCR (ddPCR) jelenleg kutatási alapon működik (14). Az SNP-táblázatokkal a PWS (az esetek kb. 85%-a) vagy AS (kb. 80%-a) jellemzőivel jelentkező betegek többségénél specifikus molekuláris osztályokat lehet azonosítani, míg a fennmaradó betegeknél további vizsgálatokra lesz szükség a grafikus kivonatban leírtak szerint. Speciális fejlett genetikai vizsgálatok (pl. ddPCR) megfelelően érzékenyek lehetnek a mozaikizmus számszerűsítésére, és a PWS és AS enyhébb klinikai jellemzőivel rendelkező egyének nagy részhalmazában azonosíthatják a diagnózist, de további kutatásokra van szükség.
A deléciós és nem deléciós státuszú PWS vagy AS betegek összehasonlításakor korai klinikai különbségeket találtak (29), beleértve a 15q11-q13 delécióval rendelkező PWS és AS betegek hipopigmentációját (30). Később magasabb verbális IQ-értékekről (31) vagy pszichózisról (32) számoltak be azoknál, akiknek az anyja UPD15-öt tartalmazott, szemben a PWS-ben szenvedő delécióval. Továbbá Butler és munkatársai (19) alacsonyabb adaptív pontszámokról és több kényszeres-kompulzív viselkedésről számoltak be a 15q11-q13 I. típusú delécióval rendelkező PWS egyéneknél az UPD15-höz képest. Zarcone és munkatársai (33) arról számoltak be, hogy a PWS-ben és a 15q11-q13 I. típusú delécióban szenvedő egyéneknek több a személyes tisztasággal kapcsolatos kényszerképzete és nehezen megszakítható és a társas tevékenységeket jobban zavaró kényszeres viselkedése, mint a II. típusú delécióban vagy az UPD15-ben szenvedőknek. Az Angelman-szindrómában végzett fenotípus-genotípus korrelációs vizsgálatban Moncla és munkatársai (34) a nagyobb I. osztályú delécióval rendelkezőknél fokozott rohamaktivitásról számoltak be a nem delécióval rendelkezőkhöz képest. Mikrokefália, ataxia, hipotónia és táplálkozási nehézségek szintén valószínűbbek a deléciós altípusban (3). Náluk súlyosabb lehet a nyelvi károsodás, különösen a receptív nyelv és az autisztikus vonások (21, 35). Az apai UPD-vel rendelkező AS-es egyéneknél javulhat a receptív nyelv, javulhatnak a motoros képességek és csökkenhet a rohamok gyakorisága. A mozaikos egyéneknek enyhébb fenotípusuk is lehet, beleértve a jobb nyelvi képességeket, az adaptív funkciókat és a kevesebb rohamot (36).
A következő generációs exom- vagy teljes genom-szekvenálásnak is helye lehet a PWS vagy AS genetikai értékelésében, különösen azoknál az egyéneknél, akiknél szokatlan lelet vagy késleltetett diagnózis (pl. UPD15 szegmentális vagy teljes izodiszómia) áll fenn, valamint olyan esetekben, amikor a szülői DNS nem áll rendelkezésre (8). A PWS és AS genetikai vizsgálatának használatára és típusára vonatkozóan a klinikusok számára egy új genetikai vizsgálati folyamatábrát dolgoztak ki, amelyet a grafikus összefoglaló ismertet és illusztrál. Ez a folyamatábra segítséget nyújthat a genetikai vizsgálat elrendelésében a klinikai megjelenés alapján a megfelelő diagnózis, kezelés és kezelés meghatározása, valamint a legpontosabb genetikai tanácsadással kapcsolatos információk biztosítása érdekében a többi családtag számára. Javasoljuk ennek az algoritmusnak a használatát mind a PWS, mind az AS esetében a diagnosztikai munka végleges befejezéséhez. Azt állítjuk, hogy a diagnózis nem teljes a páciens specifikus genetikai altípusának ismerete nélkül, amely a tanácsadás, az előrejelző iránymutatás, a kezelés és a valószínűsíthető terápiás lehetőségek irányításához szükséges. A molekuláris osztály meghatározása fontos az orvosi ellátás és kezelés szempontjából, és hasznos a PWS vagy AS családtagok genetikai tanácsadásában részt vevő klinikus számára.
Author Contributions
MB és JD hozzájárultak a kézirat megszövegezéséhez, az irodalom áttekintéséhez, szakértelmükkel hozzájárultak és szerkesztették a kéziratot.
Finanszírozás
Elismerjük a National Institute of Child Health and Human Development HD02528 számú támogatását.
Érdekellentét
A szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségként értelmezhetők.
Köszönet
Megköszönjük Grace Grahamnek a kézirat szakértői elkészítését.
1. Bittel DC, Butler MG. Prader-Willi-szindróma: klinikai genetika, citogenetika és molekuláris biológia. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. A Prader-Willi-szindróma kezelése. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Az Angelman-szindróma klinikai és genetikai vonatkozásai. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Az Angelman-gyulladásos betegségben szenvedő betegeknek a betegséggel összefüggő tünetegyüttese. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willi-szindróma. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Prader-Willi-szindróma: a klinikai, genetikai és endokrinológiai leletek áttekintése. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. PubMed Abstract | CrossRef Full Text | Google Scholar
. Butler MG. Az elhízás egygénes és szindrómás okai: szemléletes példák. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. A Prader-Willi-szindróma imprinting-központjának elemzése cseppnyi digitális PCR és újgenerációs teljes exom szekvenálás segítségével. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Három, imprinting center mikrodeléciók által okozott Prader-Willi-szindrómás testvérpár és áttekintés. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Am J Med Genet A. (2018) 176:886-95. Hassan M, Butler MG. Prader-Willi-szindróma és atipikus szubmikroszkópos 15q11-q13 deléciók imprinting-hibával vagy anélkül. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Hosszú távú kutatások a gyermekkorban és a családban. Fountain MD, Schaaf CP. Prader-Willi-szindróma és Schaaf-Yang-szindróma: a MAGEL2 génnél kereszteződő idegrendszeri betegségek. Betegségek. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. 12. A gyermekkori betegségek és a gyermekkori betegségek vizsgálata. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. A HBII-85 C/D box kis nukleoláris RNS-klaszter apai hiánya által okozott Prader-Willi fenotípus. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Vizsgálati eredmények és eredmények, amelyek alapján a betegséget az emberi egészségre gyakorolt káros hatások és az egészségre gyakorolt káros hatásokkal összefüggésbe lehet hozni. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-szerű fenotípus, amelyet egy atipikus 15q11.2 mikrodeléció okoz. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Google Scholar
. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. A 15. kromoszóma deléciói mint a Prader-Willi-szindróma oka. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM19810205303040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. A 15-ös kromoszóma deléciójának szülői eredete Prader-Willi-szindrómában. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Willi-Willi-Willi-Willi-Willi-Willi-Willi. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Molekuláris genetikai osztályozás a Prader-Willi-szindrómában: egy több helyszínen végzett kohorszvizsgálat. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Jmedgenet 2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
. Butler MG, Thompson T. Prader-Willi-szindróma: klinikai és genetikai eredmények. Endokrinológus. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Teljes szöveg | Google Scholar
18. Butler MG. Prader-Willi-szindróma: az okok és a diagnózis jelenlegi ismerete. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19.
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Viselkedésbeli különbségek Prader-Willi-szindrómás, I. vagy II. típusú delécióval és anyai disomiával rendelkező személyek között. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. PhD. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. 21. Am J Med Genet A. (2011) 155:1040-9. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Angelman-szindróma. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Az Angelman-elmezes betegségben szenvedő betegeket és a betegségben szenvedő betegeket és a betegségben szenvedő betegeket és a betegségben szenvedő betegeket és a betegségben szenvedő betegeket. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a case-controlled study. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Hírlevél. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Clinical utility gene card for: Angleman-szindróma. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. 24. Pelc K, Cheron G, Dan B. Viselkedés és neuropszichiátriai manifesztációk Angelman-szindrómában. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Am J Med Genet A. (2020) 182:53-63. o. Buiting K, Williams C, Horsthemke B. Angelman-szindróma – betekintés egy ritka neurogenetikai rendellenességbe. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. PhD. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypical Angelman syndrome due to a mosaic imprinting defect: case reports and review of the literature. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neuroterápia. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Az Angelman-kórképek és az Angelman-kórképek vizsgálata. Butler MG, Meaney FJ, Palmer CG. Klinikai és citogén felmérés 39 Prader-Labhart-Willi-szindrómás egyénről. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.132023030307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hipopigmentáció: a Prader-Labhart-Willi-szindróma gyakori jellemzője. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. A Prader-Willi-szindróma intellektuális jellemzői: genetikai altípusok összehasonlítása. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. J Intellect Disabil Res. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Pszichotikus betegség Prader-Willi-szindrómában szenvedőknél a 15. kromoszóma anyai egyszülött diszómia miatt. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. 33. Az orvosi szakirodalom és a pszichiátriai kutatások, valamint a pszichiátriai és a pszichiátriai szakirodalom (2002) 359:135-6. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. A kényszeres viselkedés és a tanulmányi teljesítmény közötti kapcsolat a Prader-Willi-szindróma három genetikai altípusában. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. J Intellekt Disabil Res. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Vö. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Új 15q11q13 deléciók azonosítása az Angelman-szindrómában array-CGH segítségével: molekuláris jellemzés és genotípus-fenotípus összefüggések. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. A megőrzött expresszív nyelv mint a mozaikos Angelman-szindróma fenotípusos meghatározója. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar