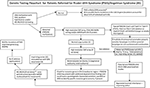
Graphical Abstract. Flussdiagramm für Gentests bei Patienten, die wegen Prader-Willi-Syndrom (PWS)/Angelman-Syndrom (AS) überwiesen werden. *Ausschluss von Chr15-Translokationen oder -Inversionen durch routinemäßige Chromosomenuntersuchungen; Berücksichtigung anderer mit Adipositas zusammenhängender genetischer Störungen; möglicherweise ist ein DNA-Screening auf FMR1-Genrepeat-Expansion (Fragiles X-Syndrom) oder eine erweiterte genetische Untersuchung mit Next-Generation-Sequencing (NGS) auf FMR1 oder andere Kandidatengenvarianten unter Verwendung von Whole-Exome-Sequencing (WES) oder Whole-Genome-Sequencing (WGS; z. B. monogene Ursachen von Adipositas) erforderlich. **Kann zur Überprüfung des Methylierungsstatus anderer Chr15-geprägter Gene verwendet werden; ddPCR, digitale Tröpfchen-PCR kann für das Mosaik-Screening verwendet werden.
Einführung
Zu den Chromosom-15-Prägungsstörungen gehören das Prader-Willi-Syndrom (PWS) und das Angelman-Syndrom (AS) (1-6) sowie Chromosom-15q-Duplikationen. Die Diagnose von PWS oder AS hängt vom Herkunftselternteil ab und davon, ob die Expression abweichend auf die mütterlichen oder väterlichen geprägten Gene beschränkt ist. Die Duplikation 15q wird durch eine zusätzliche Kopie der mütterlicherseits abgeleiteten Region 15q11.2-q13 verursacht, die zu Krampfanfällen, kognitiven und Verhaltensproblemen einschließlich Autismus-Spektrum-Störung (ASD), aber nicht zu einem PWS- oder AS-Phänotyp führen kann. PWS entsteht durch den Verlust von mütterlich geprägten und väterlich exprimierten Genen in der Region des Chromosoms 15q11-q13, während AS durch den Verlust von geprägten und mütterlich exprimierten Genen in dieser Region verursacht wird, wobei insbesondere das UBE3A-Gen betroffen ist. Aufgrund der geprägten Natur der verantwortlichen Gene können sowohl genetische als auch epigenetische Fehler ursächlich sein.
Im Jahr 1989 wurde bei Personen mit PWS und ohne Deletionsstatus eine mütterliche Disomie 15 oder beide 15 von der Mutter festgestellt, wenn polymorphe DNA-Marker aus der proximalen Region 15q11-q13 verwendet wurden (7). Später, Mitte der 1990er Jahre, wurde die Entwicklung von DNA-Sonden für die fluoreszierende In-situ-Hybridisierung (FISH) genutzt, um Deletionen der 15q11-q13-Region sowohl bei PWS als auch bei AS zu identifizieren. In diesem Zeitraum wurden auch Methylierungs-DNA-Tests entwickelt, und bei PWS und AS wurde ein abnormales Methylierungsmuster festgestellt. Methylierungs-DNA-Tests haben eine Genauigkeit von 99 % bei der Diagnose von PWS, können aber nicht die individuelle molekulare Klasse von PWS bestimmen (2). Bei AS identifiziert der DNA-Methylierungstest ~80 % der Personen, unterscheidet aber auch hier nicht zwischen den molekularen Klassen und erkennt auch keine Mutation im UBE3A-Gen, die AS verursacht.
Die Microarray-Technologie wurde Anfang bis Mitte der 2000er Jahre entwickelt und hat die diagnostische Ausbeute verbessert. Die neuen SNP-Microarrays umfassen nun über zwei Millionen DNA-Sonden und sind nützlich für den Nachweis von Deletions-Subtypen und UPD15-Unterklassen. Andere Technologien wie die digitale Tröpfchen-PCR (ddPCR) quantifizieren die Kopienzahl unter Verwendung von DNA-Sonden für Chromosom 15 und können genetische Defekte bei PWS oder AS diagnostizieren (8). Darüber hinaus können SNP-Microarrays LOHs identifizieren, die als >8 Mb groß definiert sind und, wenn sie auf Chromosom 15 vorhanden sind, die Diagnose einer mütterlichen Disomie 15 oder einer väterlichen Disomie 15 bei Vorliegen eines abnormalen DNA-Methylierungsmusters für PWS bzw. AS unterstützen. Die Bestätigung eines Imprinting-Defekts erfordert möglicherweise nicht nur SNP-Mikroarrays, um kleine Mikrodeletionen zu identifizieren, sondern auch elterliche DNA-Proben mit Genotypisierung, um das Vorhandensein einer normalen (biparentalen) Vererbung von Chromosom 15s festzustellen, was das Vorhandensein eines Epimutations-Imprinting-Defekts bei PWS oder AS unterstützt und sich somit auf das Rückfallrisiko auswirkt. Die Unterscheidung zwischen einer IC-Mikrodeletion und einem Epimutationsstatus ohne Deletion ist für Familien klinisch wichtig, da ein 50%iges Rückfallrisiko für weitere Kinder besteht, wenn bei einem Elternteil eine IC-Mikrodeletion gefunden wird (9).
Es gibt mehr als ein Dutzend Gene und Transkripte in der Region 15q11-q13, die eine Rolle bei der Entstehung von PWS und/oder AS zu spielen scheinen. Zu den Genen und Transkripten im Bereich des proximalen 15q11.2-Bruchpunkts BP1 und des distalen 15q13-Bruchpunkts BP3 gehören TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, nicht codierende RNAs (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 und HERC2. Geprägte MRKN3-, MAGEL2-, NDN-, NIPAP1- und SNURF-SNRPN-Gene werden väterlicherseits exprimiert und können, wenn sie gestört sind, Merkmale von PWS hervorrufen. So können beispielsweise Mutationen des MAGEL2-Gens zu neonataler Hypotonie, Entwicklungsverzögerung, Arthrogrypose, autistischen Merkmalen, schlechtem Saugverhalten und Fettleibigkeit führen. Es wurde auch von Patienten mit PWS-Merkmalen berichtet, die kleine Deletionen des nicht kodierenden SNORD116-Transkripts (12) und andere ähnliche Deletionen in der Region aufweisen (10, 13).
Wir haben uns in diesem Bericht auf AS und PWS konzentriert, da beide Syndrome mit Hilfe von DNA-Methylierungstests nachgewiesen werden können, was die Bestimmung des aktiven elterlichen Genallels und die endgültige Diagnose bei Personen mit PWS und bei den meisten Personen mit AS ermöglicht (2). Mit einem DNA-Methylierungstest lässt sich jedoch die molekulare Klasse bei beiden Syndromen nicht bestimmen. Die hochauflösende Chromosomenanalyse wurde in den frühen 1980er Jahren entwickelt und eingesetzt und wurde zu einem genetischen Standardtest im Labor, um die Deletion des Chromosoms 15q11-q13 zu ermitteln, die damals bei der Mehrheit der PWS-Patienten (14) und später bei AS festgestellt wurde. Über den väterlichen Ursprung der 15q11-q13-Deletion wurde 1983 berichtet (15), und es wurde festgestellt, dass es sich um eine Deletion de novo handelt, aber die Größe der 15q11-q13-Deletion oder der Typ (typisch vs. atypisch) konnten nicht bestimmt werden. Eine genaue und frühzeitige Diagnose mit Bestimmung der molekularen Klasse ist nicht nur für die Bestätigung der klinischen Diagnose, sondern auch für die genetische Beratung, die Information über Pflege und Behandlung und die Festlegung von Erwartungen von wesentlicher Bedeutung. Mit Blick auf die laufenden klinischen Studien könnte sich ein besseres Verständnis der molekularen Ätiologie auf die Möglichkeiten der Patientenbeteiligung auswirken. Zu den bevorstehenden Studien gehören auch Antisense-Oligonukleotide zur Reaktivierung der zum Schweigen gebrachten väterlichen Kopie des Chromosoms 15 bei Personen mit AS.
PWS und AS sind komplexe, seltene neurologische Entwicklungsstörungen, die auf Fehler in der genomischen Prägung zurückzuführen sind. PWS gilt als die häufigste genetische Ursache für lebensbedrohliche Fettleibigkeit, wenn sie unkontrolliert bleibt (2, 4, 6). Es gibt drei anerkannte molekulare Klassen von PWS, darunter eine väterliche 15q11-q13-Deletion mit einer Größe von etwa 5-6 Mb (60 % der Fälle) und eine mütterliche Disomie 15 (UPD15), bei der beide Chromosomen 15 von der Mutter vererbt werden (36 %), die aus einer Trisomie 15 mit Verlust des väterlichen Chromosoms 15 in der Frühschwangerschaft resultiert, was zu zwei Chromosomen 15 bei der Mutter führt (16). Bei der dritten Klasse handelt es sich um einen Defekt des Prägezentrums. Liegt auf dem väterlichen Allel eine Mikrodeletion oder Epimutation des Imprinting-Zentrums (IC) vor, das den Expressionsstatus ausgewählter geprägter Gene auf Chromosom 15 steuert, kommt es zu PWS. Dieser Imprinting-Defekt tritt bei 4 % der Personen mit PWS auf (8, 16). Die meisten Fälle von PWS treten sporadisch auf, wobei eine ungefähre Gleichverteilung nach ethnischen Gruppen und Geschlecht besteht. Die geschätzte Prävalenz von PWS liegt bei einem von 10.000 bis einem von 30.000 (2). Man geht davon aus, dass weltweit etwa 400.000 Menschen mit PWS leben, davon etwa 20.000 in den USA (2, 17).
PWS ist gekennzeichnet durch infantile Hypotonie, einen schlechten Saugreflex mit Fütterungsschwierigkeiten, Kleinwuchs mit kleinen Händen und Füßen, Hypogonadismus als Folge von Hormonmangel, leichte intellektuelle Behinderung, Verhaltensprobleme und Hyperphagie, die häufig zwischen dem sechsten und achten Lebensjahr einsetzt und bis ins Erwachsenenalter anhält und zu Fettleibigkeit führt, wenn keine Umweltkontrollen vorhanden sind. Im Säuglingsalter zeigen sich charakteristische kraniofaziale Merkmale wie ein schmaler bifrontaler Durchmesser, Schielen, eine kleine, nach oben gebogene Nase mit einer dünnen Oberlippe, nach unten gebogene Mundwinkel, klebriger Speichel und Zahnschmelzhypoplasie (2, 4, 6, 18). Die Kognition ist im Allgemeinen aufgrund des familiären Hintergrunds eingeschränkt, und zu den Verhaltensproblemen, die in der Kindheit beginnen, gehören Selbstverletzungen (Rupfen der Haut), Wutausbrüche, Sturheit und Wutanfälle mit psychiatrischen Problemen, die während dieser Zeit oder später im Jugend- oder jungen Erwachsenenalter auftreten (2). Zu den Verhaltensproblemen gehören Angstzustände, Stimmungsstörungen, Psychosen und Autismus, die mit bestimmten genetischen Subtypen oder Molekülklassen des PWS korrelieren können (19).
Historisch gesehen wird das PWS in zwei klinische Stadien eingeteilt, wobei die Gedeihstörung im Säuglingsalter das erste klinische Stadium darstellt und die Hyperphagie mit dem Auftreten von Fettleibigkeit das zweite Stadium (2). Später wurden für diese mit Adipositas zusammenhängende genetische Störung auch Ernährungsphasen beschrieben, darunter: Phase 0 mit verminderter fetaler Bewegung und Wachstumsretardierung in utero, gefolgt von Phase 1 mit Hypotonie, Gedeihstörung und Schwierigkeiten bei der Nahrungsaufnahme, Phase 2, die im Alter von etwa 2 Jahren beginnt, wenn die erste Gewichtszunahme festgestellt wird, und Phase 3, in der mangelndes Sättigungsgefühl mit Nahrungssucht und Hyperphagie einhergeht, die zu Fettleibigkeit führt, wenn sie nicht von außen kontrolliert wird. Phase 3 beginnt im Alter von etwa 6-8 Jahren (20).
Das Angelman-Syndrom ist gekennzeichnet durch eine Entwicklungsverzögerung, die oft erst im Alter von etwa 6 Monaten auftritt, und das anschließende Auftreten von oft schwer kontrollierbaren Krampfanfällen, Tremor, breitbeinigem Gang und Ataxie mit einem charakteristischen fröhlichen Verhalten (3). Es gibt vier anerkannte molekulare Mechanismen der AS: de novo mütterliche Deletionen des Chromosoms 15q11-q13 (70-80%); Mutationen des mütterlicherseits vererbten UBE3A-Gens (10-20%); väterliche Disomie 15 (3-5%); und Imprinting-Defekte (3-5%) innerhalb der Region 15q11-q13, die die Expression des ursächlichen UBE3A-Gens verändern (21).
Individuen mit AS werden von medizinischem Fachpersonal oft erst im Alter von etwa 6 Monaten bemerkt, wenn Entwicklungsverzögerungen, insbesondere eine verzögerte motorische Entwicklung, festgestellt werden. Zu diesem Zeitpunkt können die Eltern das fröhliche Verhalten erkennen, das häufiges Lachen, Lächeln und Erregbarkeit einschließt. Bei >80 % der Personen mit AS wird ein vermindertes Schlafbedürfnis festgestellt (22). Sie entwickeln häufig Anfälle im Alter von 1-3 Jahren (23). Die Epilepsie kann hartnäckig sein und hat ein charakteristisches Erscheinungsbild im EEG, das als erhöhte Delta-Energie mit einer charakteristischen dreiphasigen Welle beschrieben wird. Personen mit AS werden als ataktisch in ihren Bewegungen und beim Gehen beschrieben (24, 25). Mikrozephalie kann sich bis zum Alter von ~2 Jahren entwickeln. Zu den stereotypen Verhaltensweisen gehört eine Vorliebe für Wasser und zerknittertes Papier, und Menschen mit AS sind charakteristischerweise nonverbal und werden als schwer geistig behindert eingestuft. Es ist jedoch bemerkenswert, dass Menschen mit AS über Fähigkeiten verfügen, die mit den derzeit verfügbaren objektiven neuropsychologischen Tests nicht gut erfasst werden. Sie verfügen über ausgeprägte Fähigkeiten im Umgang mit elektronischen Geräten, aber ihr Verhalten kann schwierig sein und Angstzustände mit kurzen Aufmerksamkeitsspannen beinhalten.
Da Patienten mit PWS oder AS je nach Molekülklasse unterschiedliche Phänotypen aufweisen können und es für beide Krankheiten potenzielle Behandlungs- und Überwachungsansätze gibt, ist ein logisches Flussdiagramm für die Anordnung von Gentests durch den diese Patienten untersuchenden Arzt erforderlich. Der Schwerpunkt unseres Berichts besteht darin, die klinischen und genetischen Befunde dieser beiden genomischen Imprinting-Störungen zu beschreiben und die im klinischen Umfeld verfügbaren Optionen für Gentests sowie die Reihenfolge zu erläutern, in der die verschiedenen Gentests am produktivsten durchgeführt werden können.
Laborgenetische Erfahrungen mit Chromosom-15-Prägungsstörungen
Prader-Willi-Syndrom
Als Beispiel für die Bedeutung von hochauflösenden SNP-Microarray-Tests wurde in den USA eine große, standortübergreifende Kohorte von 510 Teilnehmern mit genetisch bestätigtem PWS rekrutiert und in drei molekulare Klassen eingeteilt. Sie wurden als 15q11-q13-Deletions-Subtypen, mütterliche Disomie-15-Subklassen und Imprinting-Center-Defekte weiter charakterisiert (16). In dieser größten berichteten PWS-Kohorte wurde bei 303 Personen (60 % der Fälle) eine 15q11-q13-Deletion festgestellt, von denen 118 Personen (38,9 %) die größere typische 15q11-q13-Deletion vom Typ I mit den Chromosomen 15q11-q13-Bruchpunkten BP1 und BP3 und 165 Personen (54.5 %) hatten die kleinere typische 15q11-q13-Deletion vom Typ II, die die Haltepunkte BP2 und BP3 einschließt. 20 Personen hatten eine atypische Deletion, die größer oder kleiner als die typische 15q11-q13-Deletion ist (6,6 %). Bei Personen, bei denen eine Deletion von Chromosom 15 festgestellt wurde, ist es wichtig zu prüfen, ob beim Vater des Probanden eine balancierte Translokation vorhanden sein könnte, da dies das Wiederholungsrisiko von PWS bei den Nachkommen des Vaters erhöht. Was die mütterliche Disomie 15 betrifft, so wiesen 185 Personen (36 %) eine mütterliche uniparentale Disomie 15 (UPD15) auf, wobei 13 Personen (12,5 %) eine totale Isodisomie des gesamten Chromosoms 15 aufgrund von Fehlern in der mütterlichen Meiose II aufwiesen; 60 (57,7 %) zeigten eine segmentale Isodisomie aufgrund von Crossover-Ereignissen in der mütterlichen Meiose I und 31 (29,8 %) eine Heterodisomie, während bei 81 Personen keine SNP-Microarray-Analyse und keine Klassifizierung der mütterlichen Disomie 15 vorgenommen wurde. Bei 22 Personen (4 %) wurden PWS-Prägungsdefekte festgestellt, wobei 13 (76,5 %) einen Nicht-Deletions-Epimutationsstatus aufwiesen, vier Personen (23,5 %) hatten eine Mikrodeletion des Prägungszentrums, während bei den übrigen fünf Personen keine Art von Prägungsdefekt festgestellt werden konnte. In einer verwandten Studie führten Hartin et al. (8) eine weitere Analyse von Imprinting-Defekten bei PWS durch, wobei sie die digitale Tröpfchen-PCR und die Ganz-Exom-Sequenzierung der nächsten Generation in einer separaten PWS-Kohorte von 15 nicht verwandten Patienten einsetzten und bei zwei Personen oder 13 % einen Mikrodeletionsdefekt des Imprinting-Zentrums fanden. Bei den 60 Personen mit segmentaler Isodisomie 15, über die Butler et al. (16) berichteten, betrug die durchschnittliche Gesamtgröße des Heterozygotieverlusts (LOH) 25,1 Mb mit einem Bereich von 5-67,4 Mb und einer durchschnittlichen Größe von 16,4 Mb für die einzelnen LOHs. Zweiunddreißig Individuen hatten ein LOH-Segment, 25 Individuen hatten zwei Segmente und drei Individuen hatten drei Segmente. Die häufigsten LOH-Stellen waren die proximale 15q11-q13-Region und die distale 15q26-Region, einschließlich der 15q12- und 15q26.1-Banden, die am häufigsten festgestellt wurden.
Das Vorhandensein einer mütterlichen UPD15 und die Bestimmung der spezifischen Unterklasse (segmentale oder totale Isodisomie) können sich auf die Diagnose und die Überwachung der medizinischen Versorgung auswirken, da eine zweite genetische Erkrankung vorliegen kann, wenn die Mutter Trägerin eines rezessiven Genallels ist, das sich in der LOH-Region befindet und zu zwei identischen Kopien führt. Hunderte von potenziell krankheitsverursachenden Genen befinden sich auf Chromosom 15, und diese Krankheiten sollten bei Personen mit segmentaler oder totaler Isodisomie von Chromosom 15 genau überprüft oder überwacht werden. Ein vorgeschlagenes Flussdiagramm für Gentests zur Identifizierung der verschiedenen molekularen Klassen sowohl für PWS- als auch für AS-Patienten ist in der grafischen Zusammenfassung zu sehen.
Angelman-Syndrom
Vier anerkannte molekulare Klassen wurden bei AS identifiziert, die nach den Auswirkungen auf die Methylierung der Chromosom-15-Region kategorisiert werden können. Der häufigste Subtyp ist eine Deletion des mütterlichen 15q11.2-q13-Region, wie sie auch bei PWS väterlicherseits vorkommt und bei ~70 % der Personen mit AS gefunden wird (21). Bei AS ist jedoch die typische Klasse-II-Deletion häufiger anzutreffen. Diese typische kleinere Klasse-II-Deletion hat meist eine Größe von ca. 5 Mb von BP2-BP3 und ist in 50 % der AS-Deletionsfälle vorhanden. Deletionen der Klasse I haben eine Größe von 5-7 Mb und umfassen BP1-BP3 (40 % der Deletionsfälle). Atypische Deletionen können sich von BP1 oder BP2-BP4 oder von weiter entfernten Bruchpunkten erstrecken. Bei Personen mit einer Deletion auf der mütterlichen Kopie von Chromosom 15 ist zu prüfen, ob auf dem Chromosomen-Mikroarray Anzeichen für Störungen zu finden sind, die auf eine mütterliche Translokation hindeuten. Dadurch erhöht sich das Risiko eines erneuten Auftretens von AS bei künftigen mütterlichen Nachkommen. Die uniparentale väterliche Disomie 15 macht 5-7 % der Personen mit AS aus. Imprinting-Defekte machen 3-5 % der Personen mit AS aus und werden durch Defekte im Imprinting-Kontrollzentrum verursacht, die von Buiting et al. zusammengefasst wurden (26). Bei Personen mit einem Defekt im Imprinting-Kontrollzentrum gelingt es der epigenetischen Markierung in der Keimbahn nicht, von einem väterlichen Muster mit unterdrückter UBE3A-Expression zu einem mütterlichen Muster der Expression des UBE3A-Gens zu wechseln. In bis zu 50 % der gemeldeten Fälle kann eine Mutation im Imprinting-Kontrollzentrum festgestellt werden. Mosaikartige Fälle von Defekten des Imprinting-Zentrums, bei denen ein Prozentsatz der Zellen keine Expression der Region 15q11.2-q13 aufweist, werden berichtet und sind möglicherweise häufiger als bisher angenommen (27). Der letzte Gendefekt bei AS wirkt sich nicht auf die Ergebnisse von DNA-Methylierungstests aus, sondern wird durch eine Mutation im mütterlicherseits vererbten UBE3A-Gen verursacht. Mutationen in diesem Gen sind für 11 % der AS-Fälle verantwortlich (28). Eine UBE3A-Mutation könnte mütterlicherseits vererbt werden, weshalb es angezeigt ist, die Mutter der Patientin gezielt zu testen, um ein 50%iges Rückfallrisiko bei ihren zukünftigen Nachkommen auszuschließen. Wenn die Mutation als vererbt angesehen wird, empfehlen wir, den Großvater mütterlicherseits zu testen, da dies Auswirkungen auf die zukünftigen Kinder der Tante mütterlicherseits haben könnte.
Diskussion
Die medizinische Behandlung von PWS und AS sollte während des Säuglingsalters von einem multidisziplinären Team geleitet werden. Sowohl Säuglinge mit PWS (häufiger) als auch mit AS können eine Gedeihstörung aufweisen. Ein Diätassistent spielt eine wichtige Rolle bei der Behandlung der Gedeihstörung und später im Kindesalter bei der Vermeidung von Fettleibigkeit durch eine eingeschränkte Ernährung und Bewegungsprogramme (ein Problem, das bei PWS häufiger auftritt, aber auch bei AS bei einigen Personen anerkannt ist). Klinische Genetiker, Orthopäden, Hausärzte, spezialisierte Ergotherapeuten (OT), Physiotherapeuten (PT) und Sprachtherapeuten (SLP), Experten für psychische Gesundheit, Schlafmediziner, Experten für psychische Gesundheit und Endokrinologen werden benötigt, um die vielfältigen Gesundheitsprobleme zu behandeln, die bei PWS auftreten können. Zu einem AS-Team gehören klinische Genetiker, Neurologen, spezialisierte Therapeuten für PT-, OT- und SLP-Dienste, Schlafspezialisten, Gastroenterologen, Physikalische Medizin und Rehabilitation, Orthopäden und Experten für psychische Gesundheit. Bei PWS ist eine angemessene medizinische Versorgung, Betreuung und Beratung erforderlich, um die Gewichtszunahme zu kontrollieren und damit verbundene Begleiterkrankungen, Verhaltensstörungen und psychiatrische Probleme zu überwachen und zu behandeln. Wachstums- und andere Hormonmängel, die bei dieser Störung häufig auftreten, müssen behandelt werden. Eine strenge Kontrolle der Ernährung mit Nahrungsmittelsicherheit und ein kontrollierter Tagesablauf mit regelmäßiger körperlicher Betätigung sind wichtige Strategien zur Kontrolle von Hyperphagie, Fettleibigkeit und damit verbundenen Komplikationen, die ein Leben lang erforderlich sind. AS erfordert ein frühzeitiges Eingreifen, einschließlich der Kenntnis spezieller therapeutischer Maßnahmen, wie z. B. unterstützende und assistierende Kommunikationsgeräte und ein Stärkungsprogramm mit intensiven Entwicklungsübungen und Aktivitäten zur Erreichung des maximalen Potenzials (z. B. SPIDER), eine frühzeitige Behandlung mit Benzodiazepinen gegen Krampfanfälle und eine Diättherapie, wie z. B. eine ketogene Diät. Die Maximierung aller Aspekte der Behandlung, einschließlich Schlafstörungen und Verstopfung, hat großen Einfluss auf die Anfallskontrolle. Ein spezialisiertes Zentrum, das mit den Feinheiten und besonderen Aspekten dieser Störungen vertraut ist, kann das Ergebnis beeinflussen.
Eine frühzeitige Diagnose ist entscheidend, um ein frühzeitiges Eingreifen sowohl bei PWS als auch bei AS zu gewährleisten. Bei PWS sollte eine frühe Diagnose im Säuglingsalter gestellt werden, um eine Wachstumshormonbehandlung einzuleiten, Ernährungsprobleme, Fettleibigkeit, Hormonmangel, Entwicklungsverzögerungen und Verhaltensprobleme zu behandeln. Die Diagnose bei AS gewährleistet auch frühzeitige Therapien, die sich auf die Entwicklungsergebnisse auswirken, sowie eine Anfallsprophylaxe einschließlich der Vorbereitung mit geeigneten Benzodiazepinen. Weitere Maßnahmen, die sich als vorteilhaft erweisen können, sind spezielle Diäten für Menschen mit AS wie die ketogene Diät oder die Therapie mit niedrigem glykämischen Index (LGIT). Eine frühzeitige Diagnose kann auch die Kosten für die medizinische Versorgung senken, indem längere Krankenhausaufenthalte aufgrund von Fütterungsproblemen bei Menschen mit PWS und Krampfanfällen bei Kindern mit AS vermieden werden.
Die Identifizierung der molekularen Klasse von PWS oder AS durch fortschrittliche genetische Tests wie hochauflösende SNP-Mikroarrays wird eine genauere Diagnose ermöglichen, die zu besseren Indikatoren für die Prognose und zu einer genaueren genetischen Beratung der Familienmitglieder führt. Hochauflösende SNP-Mikroarrays, FISH-Analysen, methylierungsspezifische Multiplex-Ligations-Sonden-Amplifikation (MS-MLPA) und/oder Genotypisierung von Chromosom 15 sind bei der Bestimmung von 15q11-q13-Deletionen nützlich. Hochauflösende SNP-Mikroarrays können die Deletions-Subtypen (typisch und atypisch) sowohl bei PWS als auch bei AS sowie die UPD15-Unterklassen (segmentale Isodisomie und totale Isodisomie) identifizieren. Der Heterodisomie-Subtyp der UPD- und IC-Defekte (Mikrodeletion und Epimutation) sowohl bei PWS als auch bei AS kann eine zusätzliche diagnostische Abklärung erfordern, wie in der grafischen Zusammenfassung dargestellt. Der Subtyp oder die Klassen haben Auswirkungen auf die Diagnose, das potenzielle Rückfallrisiko für Familienmitglieder, die Prognose und die Überwachung auf andere genetische Erkrankungen und Risikomerkmale im Zusammenhang mit der molekularen Klasse. So treten beispielsweise autistische Merkmale und Psychosen häufiger bei Menschen mit PWS und mütterlicher Disomie 15 auf und können mit den spezifischen UPD15-Unterklassen zusammenhängen. Bei Personen mit den größeren Klasse-I-Deletionen bei AS ist die Wahrscheinlichkeit größer, dass sie schwer zu behandelnde Krampfanfälle und Mikrozephalie entwickeln.
Ein Flussdiagramm für Gentests mit den verfügbaren Testoptionen, einschließlich derjenigen, die in der Vergangenheit sowohl für PWS als auch für AS verwendet wurden, ist in der grafischen Zusammenfassung aufgeführt. Die Testung auf PWS oder AS beginnt oft mit der DNA-Methylierung und geht, falls abnormal, zu anderen genetischen Testmethoden über, einschließlich hochauflösender SNP-Mikroarrays oder MS-MLPA-Assays, je nach Verfügbarkeit für die Kliniker und Familien in ihrem klinischen Umfeld. Vorzugsweise sollte ein hochauflösender SNP-Array bestellt werden, der in der westlichen medizinischen Versorgung leicht und kommerziell verfügbar ist. Die Sequenzierung der nächsten Generation (Next Generation Sequencing, NGS) des Exoms (oder des gesamten Genoms) ist für Kliniker ebenfalls verfügbar, aber die digitale Tröpfchen-PCR (ddPCR) ist derzeit forschungsbasiert (14). SNP-Arrays können bei der Mehrzahl der Patienten, die Merkmale von PWS (ca. 85 % der Fälle) oder AS (ca. 80 %) aufweisen, spezifische Molekülklassen identifizieren, während bei den übrigen Patienten zusätzliche Tests erforderlich sind, wie in der grafischen Zusammenfassung beschrieben. Spezifische fortgeschrittene genetische Tests (z. B. ddPCR) könnten ausreichend empfindlich sein, um den Mosaizismus zu quantifizieren und eine Diagnose bei einer großen Untergruppe von Personen mit milderen klinischen Merkmalen von PWS und AS zu stellen, aber es sind weitere Untersuchungen erforderlich.
Frühe klinische Unterschiede wurden beim Vergleich von PWS- oder AS-Patienten mit Deletionsstatus und solchen ohne Deletionsstatus (29) festgestellt, einschließlich Hypopigmentierung bei PWS- und AS-Patienten mit 15q11-q13-Deletion (30). Später wurde über höhere verbale IQ-Werte (31) oder Psychosen (32) bei Personen mit mütterlicher UPD15 im Vergleich zur Deletion bei Personen mit PWS berichtet. Darüber hinaus berichteten Butler et al. (19) über niedrigere adaptive Werte und mehr zwanghaftes Verhalten bei PWS-Personen mit der Deletion 15q11-q13 Typ I im Vergleich zu UPD15. Zarcone et al. (33) berichteten, dass Personen mit PWS und der Deletion 15q11-q13 Typ I mehr Zwänge in Bezug auf persönliche Sauberkeit und zwanghaftes Verhalten zeigten, das nur schwer zu unterbrechen war und soziale Aktivitäten stärker beeinträchtigte als bei Personen mit Deletionen Typ II oder UPD15. In einer Studie zur Korrelation von Phänotyp und Genotyp beim Angelman-Syndrom berichteten Moncla et al. (34) über eine erhöhte Anfallsaktivität bei Personen mit der größeren Klasse-I-Deletion im Vergleich zu Personen ohne Deletion. Mikrozephalie, Ataxie, Hypotonie und Schwierigkeiten bei der Nahrungsaufnahme sind beim Deletionssubtyp ebenfalls wahrscheinlicher (3). Sie können schwerwiegendere Sprachstörungen aufweisen, insbesondere rezeptive Sprache und autistische Züge (21, 35). Personen mit AS mit väterlicher UPD können eine verbesserte rezeptive Sprache, verbesserte motorische Fähigkeiten und eine geringere Prävalenz von Krampfanfällen aufweisen. Mosaik-Individuen können auch einen milderen Phänotyp aufweisen, einschließlich verbesserter Sprachfähigkeiten, adaptiver Funktionen und weniger Anfälle (36).
Die Exom- oder Ganzgenom-Sequenzierung der nächsten Generation kann ebenfalls einen Platz bei genetischen Evaluierungen von PWS oder AS haben, insbesondere bei Individuen mit ungewöhnlichen Befunden oder verzögerter Diagnose (z. B. UPD15 segmentale oder totale Isodisomie) und in Fällen, in denen keine elterliche DNA verfügbar ist (8). Um den Einsatz und die Art der Gentests für PWS und AS zu erörtern, wurde ein neues Flussdiagramm für Gentests für Kliniker entwickelt, das in der grafischen Zusammenfassung beschrieben und illustriert ist. Dieses Flussdiagramm kann bei der Anordnung von Gentests auf der Grundlage der klinischen Präsentation helfen, um eine geeignete Diagnose, Behandlung und Therapie zu bestimmen und die genauesten genetischen Beratungsinformationen für andere Familienmitglieder zu liefern. Wir schlagen vor, diesen Algorithmus zu verwenden, um die Diagnose sowohl für PWS als auch für AS abschließend zu klären. Wir argumentieren, dass die Diagnose ohne die Kenntnis des spezifischen genetischen Subtyps des Patienten unvollständig ist, um die Beratung, die vorausschauende Betreuung, das Management und die wahrscheinlichen therapeutischen Optionen anzuleiten. Die Bestimmung der molekularen Klasse ist wichtig für die medizinische Versorgung und Behandlung und hilfreich für den Kliniker, der Familienmitglieder mit PWS oder AS genetisch berät.
Beiträge der Autoren
MB und JD trugen zum Entwurf des Manuskripts bei, sichteten die Literatur, trugen ihr Fachwissen bei und redigierten das Manuskript.
Finanzierung
Wir erkennen die Zuschussnummer HD02528 des National Institute of Child Health and Human Development an.
Interessenkonflikt
Die Autoren erklären, dass die Forschung in Abwesenheit jeglicher kommerzieller oder finanzieller Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Anerkennungen
Wir danken Grace Graham für die fachkundige Vorbereitung des Manuskripts.
1. Bittel DC, Butler MG. Prader-Willi-Syndrom: klinische Genetik, Zytogenetik und Molekularbiologie. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Management of Prader-Willi Syndrome. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Klinische und genetische Aspekte des Angelman-Syndroms. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willi syndrome. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Prader-Willi-Syndrom: eine Übersicht über klinische, genetische und endokrine Befunde. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Einzelgene und syndromale Ursachen der Fettleibigkeit: illustrative Beispiele. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analyse des Prader-Willi-Syndroms Imprinting Center mit Droplet Digital PCR und Next-Generation Whole-Exome Sequencing. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Drei Geschwister mit Prader-Willi-Syndrom, verursacht durch Imprinting-Zentrum-Mikrodeletionen und Rückblick. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Prader-Willi-Syndrom und atypische submikroskopische 15q11-q13-Deletionen mit oder ohne Imprinting-Defekte. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Prader-Willi-Syndrom und Schaaf-Yang-Syndrom: Neuroentwicklungskrankheiten, die sich am MAGEL2-Gen kreuzen. Diseases. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi-Phänotyp verursacht durch väterlichen Mangel für den HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-Like phenotype caused by an atypical 15q11.2 microdeletion. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deletionen von Chromosom 15 als Ursache des Prader-Willi-Syndroms. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Elterlicher Ursprung der Deletion von Chromosom 15 beim Prader-Willi-Syndrom. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Molekulargenetische Klassifizierung des Prader-Willi-Syndroms: eine multisite Kohortenstudie. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Prader-Willi-Syndrom: klinische und genetische Befunde. Endocrinologist. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Prader-Willi-Syndrom: Aktuelles Verständnis von Ursache und Diagnose. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a case-controlled study. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Clinical utility gene card for: Angleman-Syndrom. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelman-Syndrom – Einblicke in eine seltene neurogenetische Störung. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypisches Angelman-Syndrom aufgrund eines Mosaik-Prägungsdefekts: Fallberichte und Überprüfung der Literatur. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Klinische und zytogene Untersuchung von 39 Personen mit Prader-Labhart-Willi-Syndrom. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hypopigmentierung: ein häufiges Merkmal des Prader-Labhart-Willi-Syndroms. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Intellektuelle Merkmale des Prader-Willi-Syndroms: Vergleich der genetischen Subtypen. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. The relationship between compulsive behavior and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preserved expressive language as a phenotypic determinant of mosaic Angelman syndrome. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar