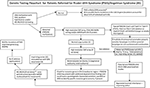
Grafický abstrakt. Schéma genetického testování u pacientů odeslaných pro Prader-Williho syndrom (PWS)/Angelmanův syndrom (AS). *Vylučte translokace nebo inverze Chr15 pomocí rutinních chromozomových studií; zvažte další genetické poruchy související s obezitou; může vyžadovat screening DNA syndromu křehkého X na expanzi opakování genu FMR1 nebo pokročilé genetické testování pomocí sekvenování nové generace (NGS) na FMR1 nebo jiné varianty kandidátních genů pomocí sekvenování celého exomu (WES) nebo sekvenování celého genomu (WGS; např. monogenní příčiny obezity). **Možno použít ke kontrole stavu metylace dalších imprintovaných genů Chr15; ke screeningu mozaicismu lze použít ddPCR, droplet digital PCR.
Úvod
Mezi poruchy imprintingu chromozomu 15 patří Prader-Williho (PWS) a Angelmanův (AS) syndrom (1-6) a duplikace chromozomu 15q. Diagnóza PWS nebo AS závisí na rodiči původu a na tom, zda je exprese aberantně omezena na mateřské nebo otcovské imprintované geny. Duplikace 15q je způsobena další kopií mateřské oblasti 15q11.2-q13, která může vést k záchvatům, kognitivním a behaviorálním problémům včetně poruch autistického spektra (ASD), ale ne k fenotypu PWS nebo AS. PWS vzniká ztrátou mateřsky imprintovaných a otcovsky exprimovaných genů z oblasti chromozomu 15q11-q13, zatímco AS je způsobena ztrátou imprintovaných a mateřsky exprimovaných genů v této oblasti, konkrétně ovlivněním genu UBE3A. Vzhledem k imprintované povaze odpovědných genů mohou být příčinou jak genetické, tak epigenetické chyby.
V roce 1989 bylo u osob s PWS i bez stavu delece při použití polymorfních markerů DNA z proximální oblasti 15q11-q13 zjištěno, že mají mateřskou disomii 15 nebo obě 15 od matky (7). Později v polovině 90. let 20. století byly díky vývoji DNA sond pro fluorescenční in situ hybridizaci (FISH) identifikovány delece oblasti 15q11-q13 jak u PWS, tak u AS. V tomto období byly vyvinuty metylační testy DNA a u PWS a AS byl pozorován abnormální metylační vzorec. Testování metylační DNA má ~99% přesnost při určování diagnózy PWS, ale neurčí jednotlivé molekulární třídy PWS (2). V případě AS metylační testování DNA identifikuje ~80 % jedinců, ale opět nerozlišuje mezi jednotlivými molekulárními třídami ani neodhalí mutaci v genu UBE3A způsobující AS.
Mikroarray technologie byla vyvinuta na počátku až v polovině roku 2000 a posunula diagnostickou výtěžnost. Nyní nové mikročipy SNP obsahují více než dva miliony sond DNA a jsou užitečné při detekci podtypů delece a podtříd UPD15. Další technologie, jako je kapková digitální PCR (ddPCR), kvantifikuje počet kopií pomocí DNA sond chromozomu 15 a může diagnostikovat genetické defekty u PWS nebo AS (8). Kromě toho lze pomocí SNP microarrays identifikovat LOH definované jako >8 Mb velké, a pokud jsou přítomny na chromozomu 15, podporují diagnózu disomie 15 u matky nebo disomie 15 u otce v přítomnosti abnormálního vzorce metylace DNA pro PWS, respektive AS. Potvrzení imprintingového defektu může vyžadovat nejen mikročipy SNP k identifikaci malých mikrodelecí, ale také vzorky rodičovské DNA s genotypizací k identifikaci přítomnosti normální (biparentální) dědičnosti chromozomu 15 podporující přítomnost epimutačního imprintingového defektu u PWS nebo AS, což ovlivňuje riziko recidivy. Odlišení mikrodelece IC od stavu bez epimutace je pro rodiny klinicky důležité, protože pokud je u rodiče zjištěna mikrodelece IC, existuje 50% riziko rekurence u dalších dětí (9).
V oblasti 15q11-q13 existuje více než tucet genů a transkriptů, které zřejmě hrají roli v příčině PWS a/nebo AS. Geny a transkripty zahrnuté v oblasti od proximálního zlomu 15q11.2 BP1 a distálního zlomu 15q13 BP3 jsou TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, nekódující RNA (SNORD), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 a HERC2. Imprintované geny MRKN3, MAGEL2, NDN, NIPAP1 a SNURF-SNRPN jsou paternálně exprimované a při narušení mohou způsobovat znaky PWS . Například mutace genu MAGEL2 mohou způsobit novorozeneckou hypotonii, opoždění vývoje, artrogrypózu, autistické rysy, špatné sání a obezitu . Byli také hlášeni pacienti s rysy PWS v důsledku malých delecí nekódujícího transkriptu SNORD116 (12) a dalších podobných delecí v této oblasti (10, 13).
V této zprávě jsme se zaměřili na AS a PWS, protože oba syndromy jsou detekovány pomocí metylačního testování DNA, které umožňuje určit aktivní alelu rodičovského genu a definitivní diagnózu u jedinců s PWS a u většiny jedinců s AS (2). Vyšetření metylace DNA však neurčí molekulární třídu ani u jednoho ze syndromů. Chromozomová analýza s vysokým rozlišením byla vyvinuta a používána na počátku 80. let 20. století a stala se standardním laboratorním genetickým testem pro hodnocení delece chromozomu 15q11-q13, která byla v té době identifikována u většiny pacientů s PWS (14) a později u AS. Otcovský původ delece 15q11-q13 byl popsán v roce 1983 (15) a bylo zjištěno, že se jedná o deleci de novo, ale velikost delece 15q11-q13 ani typ (typická vs. atypická) se nepodařilo určit. Přesná a včasná diagnóza s určením molekulární třídy je nezbytná nejen pro potvrzení klinické diagnózy, ale také pro genetické poradenství, pro informování o péči a léčbě a pro orientaci v očekáváních. Se záměrem probíhajících klinických studií může lepší pochopení molekulární etiologie ovlivnit možnosti účasti pacientů. Kromě toho se chystají studie zahrnující antisense oligonukleotidy k reaktivaci umlčené otcovské kopie chromozomu 15 u jedinců s AS.
PWS a AS jsou komplexní vzácné neurovývojové poruchy způsobené chybami v genomickém imprintingu. PWS je považována za nejčastější genetickou příčinu život ohrožující obezity, pokud není kontrolována (2, 4, 6). Existují tři uznávané molekulární třídy PWS, včetně otcovské delece 15q11-q13 o velikosti asi 5-6 Mb (60 % případů) a mateřské disomie 15 (UPD15), při níž jsou oba chromozomy 15 zděděny od matky (36 %), pocházející z trizomie 15 se ztrátou otcovského chromozomu 15 v časném těhotenství, což vede ke dvěma chromozomům 15 od matky (16). Třetí třídou je defekt imprintingového centra. Pokud je na otcovské alele přítomna mikrodelece nebo epimutace imprintingového centra (IC), které řídí stav exprese vybraných imprintovaných genů na chromozomu 15, vzniká PWS. Tento imprintingový defekt se vyskytuje u 4 % jedinců s PWS (8, 16). Většina případů PWS je sporadická s přibližnou rovností mezi etnickými skupinami a pohlavím. Odhadovaná prevalence PWS je jedna ku 10 000 až jedna ku 30 000 (2). Předpokládá se, že počet jedinců s PWS na celém světě je ~400 000, přičemž v USA žije přibližně 20 000 jedinců (2, 17).
PWS je charakterizována dětskou hypotonií, špatným sacím reflexem s obtížemi při krmení, nízkým vzrůstem s malými rukama a nohama, hypogonadismem sekundárně způsobeným nedostatkem hormonů, lehkým mentálním postižením, problémy s chováním a hyperfagií často s nástupem mezi 6. a 8. rokem věku, která přetrvává do dospělosti a vede k obezitě, pokud není zavedena kontrola prostředí. V kojeneckém věku jsou patrné charakteristické kraniofaciální rysy včetně úzkého bifrontálního průměru, strabismu, malého ohrnutého nosu s tenkým horním rtem a dolů ohrnutými koutky úst, lepkavých slin a hypoplazie skloviny (2, 4, 6, 18). Poznávací schopnosti jsou obecně snížené v závislosti na rodinném zázemí a problémy s chováním začínající v dětství zahrnují sebepoškozování (dloubání v kůži), výbuchy vzteku, tvrdohlavost a záchvaty vzteku, přičemž psychiatrické problémy se objevují v tomto období nebo později v adolescenci či mladé dospělosti (2). Behaviorální problémy zahrnují úzkost, poruchy nálady, psychózy a autismus, které mohou korelovat se specifickými genetickými podtypy nebo molekulárními třídami PWS (19).
Historicky se PWS dělí na dvě klinická stadia, přičemž neprospívání v kojeneckém věku představuje první klinické stadium a hyperfagie s nástupem obezity představuje druhé stadium (2). Později byly u tohoto genetického onemocnění spojeného s obezitou popsány další nutriční fáze, které zahrnují: Fáze 0 se sníženými pohyby plodu a růstovou retardací v děloze, následovaná fází 1 spojenou s hypotonií, neprospíváním s obtížemi při krmení, fází 2 začínající ve věku ~ 2 let, kdy je poprvé zaznamenán přírůstek hmotnosti, a fází 3, kdy je nedostatek sytosti doprovázen vyhledáváním potravy a hyperfagií vedoucí k obezitě, pokud není kontrolována zvenčí. Fáze 3 začíná přibližně ve věku 6-8 let (20).
Angelmanův syndrom je charakterizován opožděním vývoje, které se často projeví až kolem 6 měsíců věku, a následným výskytem často obtížně kontrolovatelných záchvatů, třesu, chůze na širokém podkladu a ataxie s charakteristickým veselým chováním (3). Jsou známy čtyři molekulární mechanismy AS: de novo mateřské delece chromozomu 15q11-q13 (70-80 %); mutace mateřsky děděného genu UBE3A (10-20 %); otcovská disomie 15 (3-5 %) a imprintingové defekty (3-5 %) v oblasti 15q11-q13, které mění expresi příčinného genu UBE3A (21).
U jedinců s AS si zdravotníci často všimnou až ve věku ~6 měsíců, kdy se projeví opoždění ve vývoji, zejména opožděný motorický vývoj. V této době mohou rodiče rozpoznat šťastné chování, které zahrnuje častý smích, úsměv a vzrušivost. Snížená potřeba spánku je uváděna u >80 % jedinců s AS (22). Ve věku 1-3 let se u nich často objevují záchvaty (23). Epilepsie může být neřešitelná a má charakteristický vzhled na EEG popisovaný jako zvýšená delta síla s charakteristickou trifázickou vlnou. Jedinci s AS jsou popisováni jako ataxičtí v pohybech a chůzi (24, 25). Mikrocefalie se může vyvinout do ~ 2 let věku. Stereotypní chování zahrnuje lásku k vodě a křehkému papíru a jedinci s AS jsou charakteristicky neverbální a jsou řazeni do kategorie těžce mentálně postižených. Je však pozoruhodné, že jedinci s AS mají schopnosti, které v současnosti dostupné objektivní neuropsychologické testy dobře nezachycují. Mají výrazné schopnosti v manipulaci s elektronikou, ale jejich chování může být náročné a zahrnuje úzkost s krátkou dobou pozornosti.
Jelikož se u pacientů s PWS nebo AS mohou vyskytovat variabilní fenotypy v závislosti na molekulární třídě a protože pro každou z nich existují potenciální léčebné a sledovací přístupy, je zapotřebí logické schéma pro objednávání genetických testů klinikem, který tyto pacienty hodnotí. V naší zprávě se zaměřujeme na popis klinických a genetických nálezů těchto dvou poruch genomického imprintingu a na ilustraci možností genetického testování, které jsou k dispozici v klinickém prostředí, a pořadí, v jakém lze jednotlivé genetické testy co nejproduktivněji získat.
Laboratorní genetické zkušenosti s poruchami imprintingu chromozomu 15
Prader-Williho syndrom
Pro příklad významu testování SNP microarray s vysokým rozlišením byla v USA získána velká multicentrická kohorta 510 účastníků s geneticky potvrzeným PWS, kteří byli rozděleni do tří molekulárních tříd. Ty byly dále charakterizovány jako podtypy delecí 15q11-q13, podtřídy mateřské disomie 15 a defekty imprintingového centra (16). V této největší zaznamenané kohortě PWS byla u 303 jedinců zjištěna delece 15q11-q13 (60 % případů), která se skládala ze 118 jedinců (38,9 %) s větší typickou delecí 15q11-q13 typu I zahrnující zlomové body 15q11-q13 BP1 a BP3 a 165 jedinců (54.5 %) mělo menší typickou deleci 15q11-q13 typu II zahrnující zlomové body BP2 a BP3, přičemž 20 jedinců mělo atypickou deleci, která je větší nebo menší než typická delece 15q11-q13 (6,6 %). U osob, u nichž byla zjištěna delece chromozomu 15, je důležité zvážit, zda se u otce probanda nemůže vyskytovat balancovaná translokace, protože ta zvyšuje riziko rekurence PWS u potomků otce. Pokud jde o mateřskou disomii 15, 185 jedinců (36 %) mělo mateřskou uniparentální disomii 15 (UPD15), přičemž 13 jedinců (12,5 %) mělo úplnou izodisomii celého chromozomu 15 v důsledku chyb v mateřské meióze II; 60 jedinců (57,7 %) vykazovalo segmentální izodisomii z křížových událostí v mateřské meióze I a 31 jedinců vykazovalo heterodisomii (29,8 %), zatímco u 81 jedinců nebyla provedena analýza SNP microarray a určena klasifikace mateřské disomie 15. Pokud jde o imprintingové defekty PWS, bylo zjištěno 22 jedinců (4 %), přičemž 13 (76,5 %) mělo status nedeleční epimutace, čtyři jedinci (23,5 %) měli mikrodeleci imprintingového centra, zatímco u zbývajících pěti jedinců nebyl typ imprintingového defektu stanoven. V související studii provedli Hartin et al. (8) další analýzu imprintingových defektů u PWS pomocí kapkové digitální PCR a celoexomového sekvenování nové generace u samostatné kohorty 15 nepříbuzných pacientů s PWS a u dvou jedinců, tj. 13 %, byl zjištěn defekt mikrodelece imprintingového centra. U 60 jedinců se segmentální izodisomií 15, o nichž referovali Butler et al. (16), byla celková průměrná velikost ztráty heterozygozity (LOH) 25,1 Mb s rozmezím 5-67,4 Mb a průměrnou velikostí 16,4 Mb pro jednotlivé LOH. Třicet dva jedinců mělo jeden segment LOH, 25 jedinců mělo dva segmenty a tři jedinci měli tři segmenty. Nejčastějšími místy LOH byla proximální oblast 15q11-q13 a distální oblast 15q26 včetně nejčastěji zaznamenaných pásů 15q12 a 15q26.1.
Přítomnost mateřské UPD15 a určení specifické podtřídy (segmentální nebo totální izodisomie) může mít vliv na diagnostiku a lékařský dohled, protože může být přítomno druhé genetické onemocnění, pokud je matka nositelkou recesivní genové alely umístěné v oblasti LOH vedoucí ke dvěma identickým kopiím. Na chromozomu 15 se nacházejí stovky genů potenciálně způsobujících onemocnění a tato onemocnění by měla být u osob se segmentální nebo totální izodisomií chromozomu 15 pečlivě kontrolována nebo sledována. Navrhované schéma genetického testování pro identifikaci různých molekulárních tříd u pacientů s PWS i AS si můžete prohlédnout v grafickém abstraktu.
Angelmanův syndrom
U AS byly identifikovány čtyři rozpoznané molekulární třídy, které lze rozdělit podle vlivu na metylaci oblasti chromozomu 15. V případě PWS se jedná o tři molekulární třídy. Nejčastějším podtypem je delece mateřské části 15q11.2-q13, která se podobně jako u PWS vyskytuje u otcovského původu a nachází se u ~ 70 % jedinců s AS (21). U AS je však typická delece II. třídy častější. Tato typická menší delece II. třídy má nejčastěji velikost přibližně 5 Mb od BP2-BP3 a je přítomna u 50 % případů delece u AS. Delece třídy I mají velikost 5-7 Mb a zahrnují BP1-BP3 (40 % případů delece). Atypické delece mohou sahat od BP1 nebo BP2-BP4 nebo vzdálenějších zlomových bodů. U jedinců s delecí na mateřské kopii chromozomu 15 je třeba zvážit, zda se na chromozomálním mikročipu nevyskytují známky vykazující poruchy, které naznačují, že by mohlo jít o mateřskou translokaci. To zvyšuje riziko rekurence AS u budoucích potomků matky. Uniparentální paternální disomie 15 představuje 5-7 % jedinců s AS. Imprintingové defekty představují 3-5 % jedinců s AS a jsou způsobeny defekty v imprintingovém kontrolním centru, které shrnuli Buiting et al (26). U jedinců s defektem v centru kontroly imprintingu se epigenetické značení v zárodečné linii nedokáže správně přepnout z otcovského vzorce s umlčenou expresí genu UBE3A, aby umožnilo mateřský vzorec exprese genu UBE3A. Až v 50 % hlášených případů může být identifikována mutace v imprintingovém kontrolním centru. Jsou hlášeny mozaikové případy defektů imprintingového centra, kdy určité procento buněk postrádá expresi v oblasti 15q11.2-q13, a mohou být častější, než se dříve předpokládalo (27). Poslední genetický defekt u AS nemá vliv na výsledky testování metylace DNA, ale je způsoben mutací v mateřsky děděném genu UBE3A. Mutace v tomto genu představují 11 % případů AS (28). Mutace genu UBE3A by se mohla dědit po matce, a proto je indikováno provést cílené testování u matky pacienta, aby se vyloučilo 50% riziko recidivy u jejích budoucích potomků. Pokud je mutace považována za dědičnou, doporučujeme zvážit testování dědečka pacientky z matčiny strany, protože by to mohlo mít důsledky pro budoucí děti tety z matčiny strany.
Diskuse
Medicínský management PWS a AS by měl být v kojeneckém věku řízen multidisciplinárním týmem. Jak u kojenců s PWS (častěji), tak u AS může docházet k neprospívání. Dietolog hraje důležitou roli v péči nejprve při řešení neprospívání a později v dětství, aby se zabránilo obezitě pomocí dietní intervence s omezením a využitím pohybových programů (což je problém zaznamenaný častěji u PWS, ale nyní je u některých jedinců rozpoznán i u AS). Kliničtí genetici, ortopedičtí specialisté, lékaři primární péče, specializovaní ergoterapeuti (OT), fyzioterapeuti (PT) a logopedi (SLP), odborníci na duševní zdraví, specialisté na spánek, odborníci na duševní zdraví a endokrinologové jsou potřební k řešení četných zdravotních problémů u PWS, které se mohou vyskytnout. Tým AS zahrnuje klinické genetiky, neurology, specializované terapeuty pro PT, OT a SLP služby, specialisty na spánek, gastroenterology, fyzikální medicínu a rehabilitaci, ortopedy a odborníky na duševní zdraví. U PWS je cílem odpovídající lékařská péče, management a poradenství, aby bylo možné kontrolovat přírůstek hmotnosti a sledovat a léčit přidružené komorbidní stavy, chování a psychiatrické problémy. Nedostatek růstových a jiných hormonů, který je u této poruchy běžný, vyžaduje léčbu. Důležitou strategií pro kontrolu hyperfagie, obezity a souvisejících komplikací, která je nutná po celý život, je důsledná kontrola stravy se zajištěním potravin a řízené běžné prostředí s pravidelným cvičením. AS vyžaduje včasnou intervenci včetně znalosti specializovaných terapeutických intervencí, jako jsou augmentativní a asistenční komunikační zařízení a posilovací program intenzivních vývojových cvičení a aktivit pro dosažení maximálního potenciálu (např. SPIDER), včasnou léčbu benzodiazepiny při záchvatech a dietní terapii, jako je používání ketogenní diety. Maximální využití všech aspektů péče včetně poruch spánku a zácpy výrazně ovlivňuje kontrolu záchvatů. Specializované centrum obeznámené se složitostmi a jedinečnými aspekty těchto poruch může ovlivnit výsledek.
Včasná diagnóza je zásadní pro zajištění včasné intervence jak u PWS, tak u AS. V případě PWS by měla být včasná diagnóza stanovena v kojeneckém věku, aby bylo možné zahájit léčbu růstovým hormonem, zvládnout problémy s krmením, obezitu, nedostatek hormonů, opoždění vývoje a problémy s chováním. Diagnóza u AS rovněž zajišťuje včasnou terapii, která má vliv na vývojové výsledky, a také profylaxi záchvatů včetně přípravy pomocí vhodných benzodiazepinů. Mezi další intervence, které se mohou ukázat jako prospěšné, patří specializované diety pro jedince s AS, jako je ketogenní dieta nebo terapie nízkým glykemickým indexem (LGIT). Včasná diagnóza může také snížit náklady na lékařskou péči tím, že zabrání delším hospitalizacím souvisejícím s problémy s krmením u jedinců s PWS a záchvaty u dětí s AS.
Identifikace molekulární třídy PWS nebo AS pomocí pokročilých genetických testů, jako jsou mikročipy SNP s vysokým rozlišením, umožní přesnější diagnózu, což povede k lepším ukazatelům prognózy a přesnějšímu genetickému poradenství pro členy rodiny. Mikročipy SNP s vysokým rozlišením, analýza FISH, amplifikace metylačně specifickou multiplexní ligační sondou (MS-MLPA) a/nebo genotypizace chromozomu 15 jsou užitečné při určování delecí 15q11-q13. Mikročipy SNP s vysokým rozlišením mohou určit podtypy delecí (typické a atypické) u PWS i AS a podtřídy UPD15 (segmentální izodisomie a totální izodisomie). Podtyp heterodisomie UPD a IC defekty (mikrodelece a epimutace) u PWS i AS mohou vyžadovat další diagnostické upřesnění, jak je znázorněno v grafickém abstraktu. Podtyp nebo třídy ovlivňují diagnózu, potenciální riziko recidivy pro rodinné příslušníky, prognózu a sledování dalších genetických stavů a vysoce rizikových znaků souvisejících s danou molekulární třídou. Například autistické rysy a psychózy jsou častější u osob s PWS a mateřskou disomií 15 a mohou souviset se specifickými podtřídami UPD15. U osob s většími delecemi I. třídy u AS se častěji objevují obtížně léčitelné záchvaty a mikrocefalie.
V grafickém abstraktu je uvedeno schéma genetického testování zahrnující dostupné možnosti testování včetně těch, které se historicky používaly jak u PWS, tak u AS. Testování PWS nebo AS často začíná metylací DNA, a pokud je abnormální, pak se postupuje k dalším metodám genetického testování včetně mikročipů SNP s vysokým rozlišením nebo testů MS-MLPA na základě dostupnosti pro lékaře a rodiny v jejich klinickém prostředí. Nejlépe by bylo objednat SNP array s vysokým rozlišením, která je snadno a komerčně dostupná v západní lékařské péči. Sekvenování nové generace (NGS) exomu (nebo celého genomu) je pro klinické lékaře také dostupné, ale kapková digitální PCR (ddPCR) je v současné době založena na výzkumu (14). SNP arrays mohou identifikovat specifické molekulární třídy u většiny pacientů, kteří vykazují znaky PWS (asi 85 % případů) nebo AS (asi 80 %), zatímco zbývající pacienti budou potřebovat další testování, jak je popsáno v grafickém abstraktu. Specifické pokročilé genetické testování (např. ddPCR) může být přiměřeně citlivé pro kvantifikaci mozaicismu a může určit diagnózu u velké podskupiny jedinců s mírnějšími klinickými rysy PWS a AS, ale je zapotřebí dalšího výzkumu.
Při porovnávání jedinců s PWS nebo AS s delečním stavem oproti jedincům bez delece byly zjištěny časné klinické rozdíly (29), včetně hypopigmentace u jedinců s PWS a AS s delecí 15q11-q13 (30). Později byly u osob s mateřskou UPD15 ve srovnání s delecí u jedinců s PWS zaznamenány vyšší hodnoty verbálního IQ (31) nebo psychózy (32). Butler et al (19) navíc zaznamenali nižší adaptivní skóre a více obsedantně-kompulzivního chování u jedinců s PWS s delecí 15q11-q13 typu I ve srovnání s UPD15. Zarcone et al (33) uvedli, že jedinci s PWS a delecí 15q11-q13 typu I měli více kompulzí s osobní čistotou a kompulzivní chování, které bylo obtížné přerušit a zasahovalo do sociálních aktivit více než jedinci s delecí typu II nebo UPD15. Ve studii korelace fenotypu a genotypu u Angelmanova syndromu Moncla et al. (34) zaznamenali zvýšenou záchvatovou aktivitu u osob s větší delecí I. třídy ve srovnání s osobami bez delece. U delečního podtypu se také častěji vyskytuje mikrocefalie, ataxie, hypotonie a potíže s krmením (3). Mohou mít závažnější poruchy řeči, zejména receptivní řeči a autistické rysy (21, 35). Jedinci s AS s otcovskou UPD mohou mít lepší receptivní řeč, lepší motorické schopnosti a nižší výskyt záchvatů. Jedinci s mozaikou mohou mít také mírnější fenotyp, včetně lepších jazykových schopností, adaptivních funkcí a menšího počtu záchvatů (36).
Exomové nebo celogenomové sekvenování nové generace může mít také své místo při genetickém hodnocení u PWS nebo AS, zejména u jedinců s neobvyklými nálezy nebo opožděnou diagnózou (např. segmentální nebo totální izodisomie UPD15) a v případech, kdy není k dispozici rodičovská DNA (8). K řešení otázky využití a typu genetického testování u PWS a AS byl pro lékaře vytvořen nový vývojový diagram genetického testování, který je popsán a znázorněn v grafickém abstraktu. Tento vývojový diagram může pomoci při objednávání genetického testování na základě klinického obrazu pro stanovení vhodné diagnózy, managementu a léčby a pro poskytnutí co nejpřesnějších informací o genetickém poradenství pro ostatní členy rodiny. Doporučujeme používat tento algoritmus k definitivnímu dokončení diagnostiky jak u PWS, tak u AS. Tvrdíme, že diagnóza je neúplná bez znalosti konkrétního genetického podtypu pacienta, podle kterého se řídí poradenství, anticipační vedení, management a pravděpodobné terapeutické možnosti. Určení molekulární třídy je důležité pro lékařskou péči a léčbu a užitečné pro lékaře zabývající se genetickým poradenstvím rodinných příslušníků pro PWS nebo AS.
Příspěvky autorů
MB a JD se podíleli na přípravě rukopisu, přehledu literatury, přispěli svými odbornými znalostmi a rukopis upravili.
Financování
Uznáváme grant Národního institutu pro zdraví dětí a lidský rozvoj číslo HD02528.
Střet zájmů
Autoři prohlašují, že výzkum byl prováděn bez jakýchkoli komerčních nebo finančních vztahů, které by mohly být chápány jako potenciální střet zájmů.
Poděkování
Poděkování patří Grace Graham za odbornou přípravu rukopisu.
1. Bittel DC, Butler MG. Prader-Williho syndrom: klinická genetika, cytogenetika a molekulární biologie. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Zprávy z konference, která se uskutečnila v roce 2005 v Praze. Butler MG, Lee PDK, Whitman BY. Management Prader-Williho syndromu. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Klinické a genetické aspekty Angelmanova syndromu. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Zprávy z konference, která se uskutečnila v roce 2010 v Praze. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Williho syndrom. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Zprávy z výzkumu, který se uskutečnil v roce 2012. Angulo MA, Butler MG, Cataletto ME. Prader-Williho syndrom: přehled klinických, genetických a endokrinních nálezů. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Srovnejte s výsledky výzkumu v roce 2015. Butler MG. Jednogenové a syndromové příčiny obezity: názorné příklady. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Jaké jsou výsledky výzkumu? Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analýza imprintingového centra Prader-Williho syndromu pomocí kapkové digitální PCR a celoexomového sekvenování nové generace. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Sborník příspěvků z konference, která se konala v roce 2019 v Praze. Hartin S, Hossain WA, Weisensel N, Butler MG. Tři sourozenci se syndromem Prader-Willi způsobeným mikrodelecemi imprintingového centra a přehled. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Srovnání s výsledky výzkumu v roce 2018. Hassan M, Butler MG. Prader-Williho syndrom a atypické submikroskopické delece 15q11-q13 s imprintingovými defekty nebo bez nich. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Zprávy z výzkumu, který se uskutečnil v roce 2016. Fountain MD, Schaaf CP. Prader-Williho syndrom a Schaaf-Yangův syndrom: neurovývojová onemocnění protínající se v genu MAGEL2. Onemocnění. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Dějiny a současnost. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Zjistěte, zda je možné, abyste se v případě, že se vám to nelíbí, obrátili proti sobě. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-Like phenotype caused by an atypical 15q11.2 microdeletion. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Klíčová slova v článku. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Delece chromozomu 15 jako příčina Prader-Williho syndromu. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Rodičovský původ delece chromozomu 15 u Prader-Williho syndromu. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Zprávy z výzkumu, který se uskutečnil v roce 1983. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Molekulárně genetická klasifikace u Prader-Williho syndromu: kohortová studie na více pracovištích. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. září 2019. Butler MG, Thompson T. Prader-Williho syndrom: klinické a genetické nálezy. Endokrinolog. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Zprávy o průběhu a výsledcích léčebné péče. Butler MG. Prader-Williho syndrom: současné poznatky o příčině a diagnostice. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Zprávy z konference, která se konala v roce 1990 v Praze. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. V roce 2004 se uskutečnil první ročník mezinárodní konference o dětském věku. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutriční fáze u Prader-Williho syndromu. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Srov. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Angelmanův syndrom. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Angelmanův syndrom. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a case-controlled study. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Klíčová slova v knize. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Clinical utility gene card for: Anglemanův syndrom. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Klíčová slova: Anglemanův syndrom. Pelc K, Cheron G, Dan B. Chování a neuropsychiatrické projevy u Angelmanova syndromu. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Zprávy z konference, která se uskutečnila v roce 2008, jsou k dispozici v angličtině. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Vydání časopisu Am J Med Genet. Buiting K, Williams C, Horsthemke B. Angelmanův syndrom – pohled na vzácnou neurogenetickou poruchu. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Angelmanův syndrom. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypický Angelmanův syndrom způsobený mozaikovým imprintingovým defektem: kazuistiky a přehled literatury. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neuroterapie. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Angelmanův syndrom. Butler MG, Meaney FJ, Palmer CG. Klinický a cytogenetický průzkum 39 jedinců s Prader-Labhart-Williho syndromem. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Srov. např. Butler MG. Hypopigmentace: společný rys Prader-Labhart-Williho syndromu. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Zprávy z výzkumu, který se uskutečnil v roce 1989. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Intelektové charakteristiky Prader-Williho syndromu: srovnání genetických podtypů. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Srov. např. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotické onemocnění u osob se syndromem Prader-Willi v důsledku uniparentální disomie chromozomu 15 u matky. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zprávy z konference, která se uskutečnila v roce 2002 v Praze. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. The relationship between compulsive behavior and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Srov. např. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Srov. např. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Klíčová slova v článku. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preserved expressive language as a phenotypic determinant of mosaic Angelman syndrome. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar
.