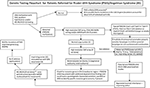
Graphical Abstract.を掲載しました。 Prader-Willi症候群(PWS)/Angelman症候群(AS)の紹介患者に対する遺伝子検査のフローチャート。 *ルーチンの染色体検査でChr15転座または逆位を除外する;他の肥満関連遺伝性疾患を考慮する;FMR1遺伝子リピート拡大のための脆弱X症候群DNAスクリーニングまたはFMR1または他の候補遺伝子変異のための次世代シーケンサー(NGS)による全エクソームシーケンサー(WES)または全ゲノムシーケンス(WGS;例えば肥満の単因性原因)による高度遺伝子検査が必要な場合もある。
はじめに
第15染色体インプリンティング障害にはPrader-Willi(PWS)症候群やAngelman(AS)症候群(1-6)、第15q重複が含まれる。 PWSやASの診断は,生みの親や,発現が母方のインプリント遺伝子に異常があるか,父方のインプリント遺伝子に異常があるかによって決まる。 15q重複は、母方由来の15q11.2-q13領域のコピーが増えることで起こり、発作、自閉症スペクトラム障害(ASD)を含む認知・行動障害を引き起こしますが、PWSやASの表現型ではありません。 PWSは染色体15q11-q13領域から母方の刷り込み遺伝子と父方の発現遺伝子が失われることによって生じ、ASはこの領域の刷り込み遺伝子と母方の発現遺伝子、特にUBE3A遺伝子に影響を与える遺伝子が失われることによって生じます。 責任遺伝子がインプリントされているため、遺伝的エラーとエピジェネティックエラーの両方が原因となりうる。
1989年に、PWSと非欠失の両方を持つ者は、近接した15q11-q13領域からの多型DNAマーカーを使用すると、母親から15番の両方または15番を持つことが判明した(7)。 その後、1990年代半ばに蛍光in situハイブリダイゼーション(FISH)DNAプローブの開発により、PWSとASの両方で15q11-q13領域の欠失を同定することができた。 この時期にメチル化DNA検査が開発され、PWSとASに異常なメチル化パターンが見られるようになった。 メチル化DNA検査は、PWSの診断の特定には〜99%の精度があるが、個々のPWSの分子クラスを特定することはできない(2)。 ASの場合、DNAメチル化検査は〜80%の個体を識別するが、やはり分子クラスを区別したり、ASを引き起こすUBE3A遺伝子の変異を検出することはできない。
マイクロアレイ技術は2000年代初期から中期に開発され、診断の歩留まりが進歩した。 現在、新しいSNPマイクロアレイは200万以上のDNAプローブを含み、欠失のサブタイプやUPD15のサブクラスの検出に有用である。 また、droplet digital PCR(ddPCR)などの技術は、15番染色体のDNAプローブを用いてコピー数を定量化し、PWSやASの遺伝子異常を診断することができる(8)。 さらに、SNPマイクロアレイは、>8 Mbの大きさと定義されるLOHを同定することができ、第15染色体に存在する場合、PWSまたはASのDNAメチル化異常パターンが存在する場合、それぞれ母方第15染色体または父方第15染色体ジソミーという診断の根拠となります。 インプリンティング異常の確認には、SNP マイクロアレイによる小さな微小欠失の同定だけでなく、親の DNA サンプルを用いて遺伝子型を調べ、PWS や AS におけるエピミューテーションインプリンティング異常の存在を裏付ける 15 番染色体の正常(両 親)遺伝の存在を確認する必要があるかもしれず、それによって再発リスクに影響することになります。 IC微小欠失と非欠失エピミューテーションの区別は家族にとって臨床的に重要であり、親にIC微小欠失が見つかった場合、追加の子供に対して50%の再発リスクが存在する(9)。 近位の15q11.2 breakpoints BP1と遠位の15q13 breakpoints BP3からの領域に含まれる遺伝子と転写物は,TUBGCP5,CYFIP1,NIPA1,NIPA2,MRKN3,MAGEL2,NDN,NIPAP1,SNURF-SNRPN,非コードRNA(SNORDs),UBE3A,ATP10A,GABRB3,GABRA5,GABRG3,OCA2およびHERC2である。 インプリンティングされたMRKN3、MAGEL2、NDN、NIPAP1、SNURF-SNRPN遺伝子は父性に発現し、障害されるとPWSの特徴を引き起こす可能性がある。 例えば、MAGEL2遺伝子の変異は、新生児低身長、発達遅延、関節隆起、自閉症的特徴、吸啜障害、肥満などを引き起こす可能性がある。 また、非コードのSNORD116転写物の小さな欠失(12)や他の類似の領域の欠失(10、13)の結果、PWSの特徴を持つ患者も報告されている。
我々はこの報告でASとPWSに焦点を当てた。 しかし、DNAメチル化検査では、どちらの症候群の分子クラスも特定することはできない。 高解像度染色体分析は1980年代初頭に開発・使用され、当時PWS患者の大半で確認されていた15q11-q13染色体の欠失(14)、後にASを評価する標準的な実験室の遺伝子ベースの検査になった。 1983年に15q11-q13欠失の父親由来が報告され(15)、de novoであることが判明したが、15q11-q13欠失のサイズやタイプ(定型と非定型)は決定できなかった。 分子クラスの特定を伴う正確な早期診断は、臨床診断の確認のみならず、遺伝カウンセリング、ケアや治療への情報提供、期待の指針として不可欠である。 現在進行中の臨床試験の意図するところでは、分子的病因のより良い理解は、患者の参加の機会に影響を与えるかもしれない。 さらに,差し迫った試験として,AS患者における15番染色体のサイレンス父方コピーを再活性化するためのアンチセンスオリゴヌクレオチドがある。
PWSとASは,ゲノム刷り込みのエラーによる複雑で稀な神経発達症である。 PWSは、コントロールされないまま放置されると生命を脅かす肥満の最も一般的な遺伝的原因として認識されている(2, 4, 6)。 PWSには、5-6Mbの大きさの父方15q11-q13欠失(60%)、妊娠初期に父方15番染色体が欠損し、母方から2本の15番染色体を受け継ぐトリソミー15に由来する母方15番ディスコミ(UPD15)(36%)という3種類の分子構造が認められている(16)。 第三は、インプリンティング・センター異常である。 15番染色体上のインプリンティング遺伝子の発現状態を制御するインプリンティング中心(IC)の微小欠失またはエピ変異が父方の対立遺伝子に存在すると、PWSが発症します。 このインプリンティングの欠陥は、PWS患者の4%に見られる(8, 16)。 PWSのほとんどは散発性で、民族や性別によっておおよその割合があります。 PWSの推定有病率は、10,000人から30,000人に1人です(2)。
PWSの特徴は、乳児期の筋緊張低下、哺乳障害を伴う吸啜反射の低下、小さな手足を伴う低身長、ホルモン欠乏による二次性性腺機能低下、軽度知的障害、行動障害、過食で、しばしば6歳から8歳の間に発症し、成人期まで続き、環境制御ができていなければ肥満になることである。 乳児期には、狭い二頭筋径、斜視、小さく上を向いた鼻、薄い上唇、下を向いた口角、粘っこい唾液、エナメル質の低形成などの特徴的な頭蓋顔面が認められます(2、4、6、18)。 認知機能は一般に家庭環境に応じて低下し、行動面では小児期から自傷行為(皮膚摘出)、暴言、頑固、癇癪などの問題があり、この時期から青年期、若年期に精神疾患が発生する(2)。
歴史的には、PWSは2つの臨床病期に分けられ、乳児期の成長不全は第1病期、肥満の発症を伴う過食は第2病期である(2)とされている。 その後、この肥満関連遺伝性疾患の栄養段階が記述されるようになり、以下のようになりました。 胎内運動低下と成長遅滞を伴う第0期、低緊張、摂食障害を伴う第1期、体重増加が初めて認められる2歳頃からの第2期、満腹感の欠如に伴う摂食障害と肥満が認められる第3期(外的コントロールを受けない場合)です。
Angelman症候群は、生後6ヶ月頃まで明らかにならない発達遅延と、その後しばしば制御困難な発作、振戦、大股歩き、運動失調が特徴的で、幸せそうな態度で発症する(3)。 ASの分子メカニズムとしては、染色体15q11-q13のde novo母体欠失(70-80%)、母方遺伝のUBE3A遺伝子の変異(10-20%)、父方の15番ディスオミー(3-5%)、15q11-q13領域内のインプリンティング欠陥(3-5%)の4つが認められており、原因となっているUBE3A遺伝子の発現を変化させる (21).
ASの患者は、特に運動発達の遅れが報告される生後6カ月頃まで医療専門家に気づかれないことが多い。 この時期までに、両親は、頻繁に笑い、微笑み、興奮を含む幸せな態度を認識することができる。 睡眠時間の短縮は、AS患者の80%において報告されている(22)。 1-3歳で発作を発症することが多い(23)。 てんかんは難治性で、脳波では特徴的な三相性波を伴うデルタパワーの増加として表現される特徴的な外観を持つ。 ASの患者は、動作や歩行に失調があると言われている(24, 25)。 小頭症は2歳までに発症することがある。 定型的な行動には、水としわくちゃの紙が好きなことが含まれ、ASの患者は特徴的に非言語的で、重度の知的障害に分類される。 しかし、ASの患者は、現在利用可能な客観的神経心理学的検査では十分にとらえられない能力を持っていることが注目される。
PWSやASの患者は分子クラスによって様々な表現型を示し,それぞれに治療や監視の可能性があるため,これらの患者を評価する臨床医が遺伝子検査をオーダーするための論理的フローチャートが必要である。 本報告では、これら2つのゲノムインプリンティング障害の臨床的・遺伝的所見を述べ、臨床現場で利用可能な遺伝子検査の選択肢と、異なる遺伝子検査を最も生産的に受けることができる順序を説明することを主眼とするものである。
Laboratory Genetics Experience in Chromosome 15 Imprinting Disorders
Prader-Willi Syndrome
高解像度SNPマイクロアレイ検査の重要性を示す例として、米国で遺伝的にPWSが確定した510名の大規模マルチサイトコホートが集められ、三つの分子クラスにグループ分けがなされた。 それらはさらに、15q11-q13欠失のサブタイプ、母体ディスオミー15のサブクラス、インプリンティングセンター欠損として特徴づけられた(16)。 この最大規模のPWSコホートでは、303人が15q11-q13欠失を有していた(症例の60%)。その内訳は、15q11-q13染色体のブレークポイントBP1とBP3を含む大きな典型的15q11-q13 Type I欠失が118人(38.9%)、165人(54.5%)が、BP2およびBP3を含むより小さな典型的な15q11-q13 Type II欠失を有し、20人が典型的な15q11-q13欠失よりも大きいか小さい非典型的欠失を有していた(6.6%)。 15番染色体の欠失が確認された場合、プロバンドの父親にバランス転座が存在するかどうかを検討することが重要です。 母性ジソミー15については、185人(36%)が母性片親性ジソミー15(UPD15)で、13人(12.5%)が母性減数第二分裂のエラーによる15番染色体全体のアイソディソミー、60人(57.7%)が母性減数第一分裂での交叉イベントによるセグメントアイソディソミー、31人がヘテロディソミーであり、81人はSNPミクロアレイ解析と母性ジソミー15分類が決定されていないことが明らかになった。 PWSのインプリンティング欠陥については、22人(4%)が発見され、13人(76.5%)が非欠損エピミューテーション状態、4人(23.5%)がインプリンティングセンターのマイクロ欠失、残りの5人はインプリンティング欠陥のタイプが確立されていなかった。 関連する研究として、Hartinら(8)は、血縁関係のない15人のPWS患者の別のコホートにおいて、液滴デジタルPCRと次世代全エクソームシーケンスを用いてPWSのインプリント欠陥についてさらなる解析を行い、2人、13%にインプリント中心のマイクロ削除の欠陥があることが明らかにされた。 Butlerら(16)が報告したセグメントイソダイソミー15を持つ60人では、ヘテロ接合性喪失(LOH)の合計平均サイズは25.1Mbで、範囲は5〜67.4Mb、個々のLOHの平均サイズは16.4Mbであった。 32人が1つのLOHセグメントを、25人が2つのセグメントを、3人が3つのセグメントを有していた。 LOH部位は近位の15q11-q13領域と遠位の15q26領域で,最も多く記録されたのは15q12と15q26.1バンドだった。
母親のUPD15の存在と特定のサブクラス(分割型または完全アイソディソミー)決定は,LOH領域にある劣性遺伝子アレルのキャリアであれば2つ同じコピーが存在することになるため,診断や医療監視に影響を与える可能性がある。 15番染色体には何百もの潜在的な疾患原因となる遺伝子が存在し、15番染色体の分節型アイソダイソミーや全アイソダイソミーを持つ人は、これらの疾患を注意深くチェックしたり監視したりする必要があるのです。
Angelman Syndrome
ASでは4つの分子クラスが確認されており、15番染色体領域のメチル化に与える影響により分類されています。 最も一般的なサブタイプは,母方の15q11の欠失である。2-q13領域は、PWSの父親由来と同様に、ASの個体の約70%に見られる(21)。 しかし、ASでは典型的なクラスII欠失がより一般的である。 この典型的な小さいクラスII欠失は、最も一般的にはBP2-BP3から約5Mbの大きさで、欠失性AS症例の50%に存在する。 Class Iの欠失は5-7Mbの大きさで、BP1-BP3を包含する(欠失症例の40%)。 非典型的な欠失はBP1またはBP2-BP4から伸びるか、あるいはもっと離れたブレークポイントから伸びることもある。 15番染色体の母方のコピーに欠失がある個体では、染色体マイクロアレイで母方の転座の可能性を示す障害があるかどうかを検討する必要があります。 これは、将来母方の子孫にASが再発するリスクを高めることになります。 父方の片親性ジソミー15は、AS患者の5~7%を占めます。 インプリンティング欠陥はAS患者の3〜5%を占め、Buitingら(26)により要約されたインプリンティングコントロールセンターの欠陥により引き起こされる。 インプリント制御センターに欠陥がある個体では、生殖細胞系列におけるエピジェネティックなマーキングは、UBE3A発現がサイレンシングされた父性パターンからUBE3A遺伝子における母性パターンの発現を可能にするために適切に切り替えることができない。 報告例の50%程度で、インプリンティング制御中枢の変異が同定されることがある。 細胞の何割かが15q11.2-q13領域の発現を欠くインプリンティングセンター欠損のモザイク症例が報告されており、以前考えられていたよりも多い可能性がある(27)。 ASの最後の遺伝的欠陥は、DNAメチル化検査結果に影響を与えないが、母方遺伝のUBE3A遺伝子の変異によって起こる。 この遺伝子の変異は、AS症例の11%を占めている(28)。 UBE3A遺伝子変異は母系遺伝する可能性があるため、患者の母親を対象とした検査を行い、将来の子孫における50%の再発リスクを排除することが望ましい。
議論
PWSとASの医学的管理は、乳幼児期に学際的なチームによって指示されるべきである。 PWS(より一般的)とASの両方の乳児は、成長不良を起こすことがある。 栄養士は、最初は成長障害に対処するために、その後は食事制限や運動プログラムの使用により肥満を回避するために、ケアにおいて重要な役割を果たす(これはPWSによく見られる懸念であるが、現在ではASでも一部の人に認められている)。 PWSで起こりうる複数の健康問題に対処するために、臨床遺伝学者、整形外科専門医、プライマリーケア医、専門の作業(OT)、理学(PT)、言語(SLP)療法士、メンタルヘルス専門家、睡眠専門家、精神保健専門家、内分泌専門家が必要とされています。 ASチームには、臨床遺伝学者、神経学者、PT、OT、SLPサービスのための専門セラピスト、睡眠専門家、消化器内科、身体医学とリハビリテーション、整形外科、精神衛生の専門家などが含まれます。 PWSの場合、適切な医療ケア、管理、カウンセリングは、体重増加をコントロールし、関連する併存疾患、行動、精神的問題を監視し治療するための目標です。 この疾患では、成長ホルモンやその他のホルモンの欠乏がよくみられるため、治療が必要である。 食の安全を確保した上で食事を厳密に管理し、定期的な運動で日常環境を管理することが、生涯を通じて必要とされる過食、肥満および関連する合併症を制御する重要な戦略である。 ASでは、補強型および補助型のコミュニケーション機器や、最大限の可能性を引き出すための集中的な発達運動や活動の強化プログラム(例:SPIDER)などの専門的な治療介入に関する知識、発作に対するベンゾジアゼピン系の早期治療、ケトジェニック食の使用などの食事療法を含む早期介入が必要である。 睡眠障害や便秘を含むあらゆるケアの最大化が発作のコントロールに大きく影響する。 これらの疾患の複雑で独特な側面に精通した専門施設であれば,転帰に影響を与えることができる。 PWSでは,成長ホルモン治療を開始し,摂食障害,肥満,ホルモン欠乏,発達遅延,行動問題を管理するために,乳児期に早期診断がなされるべきである。 また、ASの診断により、発達に影響する早期治療や、適切なベンゾジアゼピン系薬剤による発作の予防が可能となる。 その他、ケトジェニック・ダイエットや低血糖指数療法(LGIT)など、AS患者向けの特殊な食事療法も有効であることが分かっている。
高分解能SNPマイクロアレイなどの高度な遺伝子検査によりPWSまたはASの分子クラスを特定することで,より正確な診断が可能となり,予後の指標となり,家族への遺伝カウンセリングもより正確に行えるようになる。 15q11-q13欠失の判定には、高解像度SNPマイクロアレイ、FISH分析、メチル化特異的多重ライゲーションプローブ増幅法(MS-MLPA)、15番染色体遺伝子型判定などが有効である。 高解像度SNPマイクロアレイは、PWSとASにおける欠失のサブタイプ(定型と非定型)、およびUPD15のサブクラス(セグメントアイソディソミーとトータルアイソディソミー)を同定することができる。 PWSとASにおけるUPDのヘテロダイソミーとIC欠損(微小欠失とエピミューテーション)のサブタイプは、図解に示すように、診断に追加作業が必要な場合がある。 サブタイプやクラスは、診断、家族への再発リスク、予後、他の遺伝性疾患や分子クラスに関連する高リスクの特徴の監視に影響を与える。 例えば、自閉症や精神病は、PWSや母体ジソミー15によく見られ、UPD15の特定のサブクラスに関連している可能性があります。 ASの大きなクラスIの欠失を持つ者は、治療が困難な発作や小頭症を発症しやすい。
PWSとASの両方に歴史的に使用されてきたものを含め、利用できる検査オプションを組み込んだ遺伝子検査のフローチャートをグラフの要旨に記載している。 PWSまたはASの検査は、しばしばDNAメチル化から始まり、異常があれば、臨床医とその家族の臨床環境における利用可能性に基づいて、高解像度SNPマイクロアレイまたはMS-MLPAアッセイなどの他の遺伝子検査法へと進む。 好ましくは、西洋医学で容易に市販されている高解像度SNPアレイを注文することであろう。 エクソーム(または全ゲノム)の次世代シーケンサー(NGS)も臨床医が利用できるが、現在はdroplet digital PCR(ddPCR)が研究ベースとなっている(14)。 SNPアレイは、PWS(症例の約85%)またはAS(約80%)の特徴を呈する患者の大部分で特定の分子クラスを特定することができるが、残りの患者はGraphical Abstractに記載されているように追加の検査が必要である。
PWSやASの15q11-q13欠失の患者における色素沈着など、欠失と非欠失の患者を比較すると、早期の臨床差が認められた(29)。 その後、PWSの欠失と比較して、母親のUPD15では言語性IQスコアが高いこと(31)や精神病(32)が報告されている。 さらに、Butlerら(19)は、15q11-q13 Type I欠失のPWS患者では、UPD15と比較して適応得点が低く、強迫観念的な行動が多いことを報告している。 Zarconeら(33)は、PWSで15q11-q13のI型欠失を持つ人は、II型欠失やUPD15を持つ人よりも、個人の清潔さに対する強迫観念や、中断が困難で社会活動を阻害する強迫行為が多いと報告している。 アンジェルマン症候群の表現型-遺伝子型相関研究において、Monclaら(34)は、非欠失に比べ、より大きなI型欠失を持つものでは発作の活性が増加すると報告している。 小頭症、運動失調、筋緊張低下、摂食障害も欠失型サブタイプで起こりやすいとされている(3)。 特に受容言語の言語障害が強く、自閉的な特徴を持つことがある(21, 35)。 父親がUPDのASの場合、受容言語の改善、運動能力の向上、発作の有病率の減少がみられることがある。 モザイク個体はまた、言語能力の向上、適応機能、および発作の減少を含むより穏やかな表現型を有するかもしれない(36)。
次世代のエクソームまたは全ゲノム配列決定は、PWSまたはASの遺伝子評価において、特に異常所見を呈する個体や診断が遅れた場合(例えば、UPD15のセグメントまたは完全アイソダイソー)、および親のDNAが得られない場合にも位置付けられる可能性がある(8)。 PWSとASの遺伝子検査の使用と種類に対処するために、臨床医のための新しい遺伝子検査フローチャートが開発され、Graphical Abstractに記載・図解された。 このフローチャートは、適切な診断、管理、治療を決定するために、臨床症状に基づいて遺伝子検査を指示し、他の家族のために最も正確な遺伝カウンセリング情報を提供するのに役立ちます。 PWSとASの診断を確定的にするために、このアルゴリズムを使用することを提案する。 我々は、カウンセリング、予期指導、管理、治療の選択肢を導くために、患者の特定の遺伝的サブタイプを知らなければ、診断は不完全であると主張する。 MBとJDは原稿の作成、文献のレビュー、専門知識の提供、原稿の編集に貢献した。
利益相反
著者らは、潜在的な利益相反と解釈される商業的または金銭的関係がない状態で研究が行われたことを宣言する。
謝辞
原稿を専門的に作成したGrace Grahamに感謝する。
1. Bittel DC, Butler MG. プラダーウィリー症候群:臨床遺伝学、細胞遺伝学、分子生物学。 Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2.プラダー・ウィリー症候群:臨床遺伝学と細胞遺伝学、分子生物学。 バトラーMG、リーPDK、ウィットマンBY. プラダーウィリー症候群のマネージメント。 ニューヨーク、NY。 Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA、Driscoll DJ、Dagli AI.は、プラダー・ウィリー症候群の治療法について研究している。 アンジェルマン症候群の臨床的・遺伝的側面。 Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4.アンジェルマン症候群の臨床と遺伝の側面。 Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willi症候群. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5.プラダー・ウィリー症候群(Prader-Willi syndrome)。 Angulo MA, Butler MG, Cataletto ME. Prader-Willi 症候群:臨床、遺伝、内分泌所見のレビュー。 J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6.プラダー・ウィリー症候群:臨床的、遺伝的、内分泌的所見のレビュー(Prader-Willi syndrome: Review of the clinical and genetic and endocrine findings). Butler MG. 肥満の単一遺伝子と症候群性原因:実例。 Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader-Willi syndrome.(非分裂性プラダーウィリー症候群における母親のヘテロダイソミーによる遺伝刷り込み. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8.遺伝的インプリンティングは、非分裂型プラダーウィリー症候群の母親のヘテロダイソミーで示唆されている。 Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. プラダーウィリー症候群インプリンティング中心の液滴デジタルPCRと次世代全エクソームシーケンスによる解析。 モル・ジェネ・ゲノミック・メッド。 (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9.プラダーウィリー症候群のインプリンティングセンターの解析。 Hartin S, Hossain WA, Weisensel N, Butler MG. インプリンティングセンター微小欠失によるPrader-Willi症候群の3兄妹とレビュー。 Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10.インプリンティング中心微小欠失によるPrader-Willi症候群の3人の同居者、およびそのレビュー。 Hassan M, Butler MG. Prader-Willi症候群と非典型的な微小な15q11-q13欠失は、インプリンティングの欠陥を伴うか伴わない。 Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11.プラダー・ウィリー症候群と典型的な亜微小な15q11-q13欠失。 Fountain MD, Schaaf CP. Prader-Willi症候群とSchaaf-Yang症候群:MAGEL2遺伝子で交差する神経発達障害。 Diseases. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text|Google Scholar
12.プラダー・ウィリー症候群とシャーフ・ヤン症候群:MAGEL2遺伝子で交差する神経発達症 Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. HBII-85 C/D box small nucleolar RNA clusterの父親欠損によるプラダーウィリー表現型。 Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13.プラダー・ウィリー表現型は、父方のHBII-85 C/Dボックス小核RNAクラスターの欠損に起因する。 Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. 非定型15q11.2微小欠失によるPrader-Willi-Like phenotype. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text|Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. 15番染色体の欠失はPrader-Willi症候群の原因である。 N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15.染色体欠損は、Prader-Willi症候群の原因として、15番染色体の欠失である。 Butler MG, Palmer CG. Prader-Willi症候群における15番染色体欠失の親由来。 Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16.プラダー・ウィリー症候群の15番染色体の欠失の両親の起源。 Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. プラダーウィリー症候群の分子遺伝学的分類:マルチサイトコホート研究。 Jメドジェネ。 (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17.プラダー・ウィリー症候群の分子遺伝学的分類:多施設コホート研究. Butler MG, Thompson T. Prader-Willi syndrome: clinical and genetic findings. Endocrinologist。 (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18.バトラーMG、トンプソンT.プラダーウィリー症候群:臨床と遺伝学的所見。 バトラーMG. プラダーウィリー症候群:原因と診断に関する現在の理解。 Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19.プラダーウィリー症候群:現在の原因と診断の理解(Prader-Willi syndrome: current understanding of the cause and diagnosis). バトラーMG、ビッテルDC、キビリエバN、タレビザデZ、トンプソンT.プラダーウィリー症候群とI型またはII型欠失と母親のディスノミを持つ被験者間の行動的差異。 小児科。 (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20.プラダーウィリー症候群とI型またはII型欠失と母体ディスノミの被験者の行動の違い。 Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Prader-Willi症候群における栄養段階。 Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21.プラダーウィリー症候群の栄養フェーズ. このように、アンジェルマン症候群の分子クラスは、表現型によって区別される。 J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22.アンジェルマン症候群の分子クラスは、それぞれ異なる表現型である。 Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Angelman症候群の子どもにおける睡眠の多方面からの評価:症例対照研究. フロント・サイキヤトリー。 (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23.アンジェルマン症候群の子どもにおける睡眠の多方面からの評価:症例対照研究(Multi-method assessment in children with Angelman syndrome). Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Clinical Utility gene card for.臨床的有用性のある遺伝子カード。 アングルマン症候群。 Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Angelman症候群における行動と精神神経症状。 Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25.アンジェルマン症候群の行動と神経精神症状。 Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. Angelman症候群の子供の大規模臨床コホートにおける健康問題と発達の概観。 Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26.アンジェルマン症候群の子供の大規模な臨床コホートにおける健康上の問題と発達の概要。 アンジェルマン症候群-まれな神経遺伝学的疾患への洞察-. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. モザイクインプリンティング欠損による非定型アンジェルマン症候群:症例報告と文献のレビュー。 Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28.邦訳:「非定型アンジェルマン症候群」。 Margolis SS, Sell GL, Zbinden MA, Bird LM. アンジェルマン症候群。 ニューロセラピューティック。 (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29.アンジェルマン症候群(Angelman syndrome). Butler MG, Meaney FJ, Palmer CG. Prader-Labhart-Willi症候群39名の臨床的および細胞遺伝学的調査。 Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30.プラダー・ラバート・ウィリー症候群の臨床的・細胞遺伝学的調査(Prader-Labhart-Willi syndrome 39人)。 Butler MG. 色素沈着:Prader-Labhart-Willi症候群の一般的な特徴。 Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31.色素沈着症:プラダー・ラバート・ウィリー症候群に共通する特徴。 Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. プラダーウィリー症候群の知能的特徴:遺伝的サブタイプの比較. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. また、このような場合、「虹の架け橋」としての役割を果たすことが期待される。 Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33.精神科医:「精神科医になるために必要なこと」、「精神科医になるために必要なこと」、「精神科医になるために必要なこと」、「精神科医になるために必要なこと」。 Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. 強迫行為と学力の関係は、プラダーウィリー症候群の3つの遺伝子サブタイプにまたがっている。 J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34.学業成績と強迫行動の関係 Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. 20 deletion and 20 non-deletion Angelman syndrome patientsにおける表現型-遺伝子型の相関性. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. アレイCGHによるアンジェルマン症候群の15q11q13の新規欠失の同定:分子特性および遺伝子型と表現型の相関関係. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36.アンジェルマン症候群の新規15q11q13欠失のアレイCGHによる同定。 Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preserved expressive language as a phenotypic determinant of mosaic Angelman syndrome(モザイク版アンジェルマン症候群の表現型決定因子としての表現力の保持)。 Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar