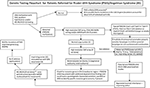
Abstract Gráfico. Fluxograma de testes genéticos para pacientes encaminhados para a síndrome de Prader-Willi (PWS)/Angelman (AS). *Regular translocações ou inversões Chr15 por estudos cromossômicos de rotina; considerar outros distúrbios genéticos relacionados à obesidade; pode requerer triagem de DNA frágil da síndrome X para expansão de repetição do gene FMR1 ou testes genéticos avançados com sequenciamento de próxima geração (NGS) para FMR1 ou outras variantes gênicas candidatas usando sequenciamento de todo o exógeno (WES) ou sequenciamento de todo o genoma (WGS; por exemplo, causas monogênicas de obesidade). **Pode ser usado para verificar o estado de metilação de outros genes impressos no Chr15; ddPCR, a PCR digital de gotículas pode ser usada para rastreio de mosaicismo.
Introdução
As doenças de impressão do cromossoma 15 incluem as síndromes Prader-Willi (PWS) e Angelman (AS) (1-6) e duplicações do cromossoma 15q. O diagnóstico de PWS ou AS depende do pai de origem e se a expressão está aberrantemente limitada aos genes maternos ou aos genes paternos impressos. A duplicação 15q é causada por uma cópia adicional da região 15q11.2-q13 derivada da maternidade que pode levar a convulsões, problemas cognitivos e comportamentais, incluindo a desordem do espectro do autismo (ASD), mas não um fenótipo de PWS ou AS. A PWS surge da perda de genes impressos e expressos por via paterna da região do cromossoma 15q11-q13, enquanto que a AS é causada pela perda de genes impressos e expressos por via materna nesta região, impactando especificamente o gene UBE3A. Devido à natureza impressa dos genes responsáveis, tanto erros genéticos quanto epigenéticos podem ser causadores.
Em 1989, foi constatado que aqueles com PWS e sem estado de eliminação tinham disomia materna 15 ou ambos 15s da mãe ao usar marcadores de DNA polimórfico da região proximal 15q11-q13 (7). Mais tarde, em meados dos anos 90, o desenvolvimento de sondas de DNA fluorescentes de hibridização in situ (FISH) foi usado para identificar deleções da região 15q11-q13 tanto na PWS quanto na AS. O teste de DNA por metilação foi desenvolvido durante este período de tempo e um padrão anormal de metilação foi observado em PWS e AS. O teste de DNA de metilação é ~99% preciso na identificação do diagnóstico de PWS, mas não identificará a classe molecular individual de PWS (2). Para AS, o teste de metilação de DNA identifica ~80% dos indivíduos, mas mais uma vez não distingue entre as classes moleculares ou detecta uma mutação no gene UBE3A causando AS.
A tecnologia Microarray foi desenvolvida no início a meados de 2000 e avançou o rendimento do diagnóstico. Agora os novos microarrays SNP incluem mais de dois milhões de sondas de DNA e são úteis na detecção de subtipos de deleção e subclasses UPD15. Outra tecnologia como a PCR digital de gotículas (ddPCR) quantifica o número de cópias usando sondas de DNA do cromossomo 15 e pode diagnosticar defeitos genéticos em PWS ou AS (8). Além disso, os microarrays SNP podem identificar LOHs definidos como >8 Mb em tamanho e quando presentes no cromossoma 15 suportam o diagnóstico da disomia materna 15 ou disomia paterna 15 na presença de um padrão anormal de metilação de DNA para PWS ou AS, respectivamente. A confirmação do defeito de impressão pode requerer não apenas microarrays SNP para identificar pequenas microdeleções, mas também amostras de DNA parental com genotipagem para identificar a presença de herança normal (biparental) do cromossomo 15, suportando a presença de um defeito de impressão de epimutação em PWS ou AS, impactando assim os riscos de recorrência. A diferenciação de uma microdeleção IC de um estado de epimutação sem epimutação é clinicamente importante para as famílias, pois um risco de recorrência de 50% está presente para crianças adicionais se uma microdeleção IC for encontrada nos pais (9).
Existem mais de uma dúzia de genes e transcrições na região 15q11-q13 que parecem desempenhar um papel na causa da PWS e/ou AS. Genes e transcrições incluídos na área dos pontos de parada proximais 15q11.2 BP1 e distal 15q13 BP3 são TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, RNAs não codificadores (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2, e HERC2. Os genes impressos MRKN3, MAGEL2, NDN, NIPAP1 e SNURF-SNRPN são expressos de forma paternal e quando perturbados podem causar características de PWS . Por exemplo, mutações do gene MAGEL2 podem causar hipotonia neonatal, atraso no desenvolvimento, artrogripose, características autistas, uma má sucção e obesidade . Os pacientes também têm sido relatados com características de PWS como resultado de pequenas deleções da transcrição SNORD116 não codificadora (12) e outras deleções similares na região (10, 13).
Centramo-nos no AS e PWS neste relatório, já que ambas as síndromes são detectadas através de testes de metilação de DNA, o que permite a determinação do alelo do gene parental ativo e o diagnóstico definitivo em indivíduos com PWS e na maioria dos indivíduos com AS (2). Entretanto, o teste de metilação de DNA não identificará a classe molecular em nenhuma das síndromes. A análise cromossômica de alta resolução foi desenvolvida e utilizada no início da década de 1980 e tornou-se um teste padrão de laboratório baseado em genética para avaliar a deleção do cromossomo 15q11-q13 identificada na maioria dos pacientes com PWS naquela época (14) e posteriormente para AS. A origem paterna da deleção 15q11-q13 foi relatada em 1983 (15) e encontrada de novo, mas o tamanho da deleção ou tipo 15q11-q13 (típica vs. atípica) não pôde ser determinado. O diagnóstico exato e precoce com identificação da classe molecular é essencial não só para confirmar o diagnóstico clínico, mas também para o aconselhamento genético, para informar os cuidados e tratamentos e para orientar as expectativas. Com a intenção de ensaios clínicos em curso, uma melhor compreensão da etiologia molecular pode impactar as oportunidades de participação do paciente. Além disso, estudos iminentes incluem oligonucleotídeos antisensos para reativar a cópia paterna silenciada do cromossomo 15 em indivíduos com AS.
PWS e AS são distúrbios neurodevelopmentais raros e complexos devido a erros na impressão genômica. PWS é reconhecida como a causa genética mais comum de obesidade potencialmente fatal, se deixada descontrolada (2, 4, 6). Existem três classes moleculares de PWS reconhecidas, incluindo uma deleção paterna 15q11-q13 de cerca de 5-6 Mb (60% dos casos) e uma disomia materna 15 (UPD15) na qual ambos os cromossomos 15 são herdados da mãe (36%) com origem na trissomia do cromossomo 15 com perda do cromossomo 15 paterno no início da gravidez levando a dois cromossomos 15 da mãe (16). A terceira classe é um defeito do centro de impressão. Se uma microdeleção ou epimutação do centro de impressão (CI), que controla o status de expressão de genes impressos selecionados no cromossomo 15, estiver presente no alelo paterno, então ocorre a PWS. Este defeito de impressão é observado em 4% dos indivíduos com PWS (8, 16). A maioria dos casos de PWS são esporádicos com uma eqüidade aproximada entre grupos étnicos e sexo. A prevalência estimada de PWS é de uma em 10.000 para uma em 30.000 (2). O número de indivíduos em todo o mundo com SPW é estimado em ~400.000, com cerca de 20.000 indivíduos vivendo nos EUA (2, 17).
A SPW é caracterizada por hipotonia infantil, um reflexo de sucção pobre com dificuldades de alimentação, baixa estatura com mãos e pés pequenos, hipogonadismo secundário a deficiências hormonais, deficiência intelectual leve, problemas comportamentais e hiperfagia, muitas vezes com início entre 6 e 8 anos de idade que persiste na idade adulta e resulta em obesidade se não houver controles ambientais. Durante a infância, são observadas características craniofaciais, incluindo um diâmetro bifrontal estreito, estrabismo, pequeno nariz virado para cima com um lábio superior fino, e cantos da boca virados para baixo, saliva pegajosa e hipoplasia do esmalte (2, 4, 6, 18). A cognição é geralmente reduzida com base nos antecedentes familiares e os problemas de comportamento que começam na infância incluem automutilação (apanhar a pele), explosões, teimosia e birras temperamentais com problemas psiquiátricos que ocorrem durante este período ou mais tarde na adolescência ou na idade adulta jovem (2). Os problemas comportamentais incluem ansiedade, distúrbios de humor, psicose e autismo que podem se correlacionar com subtipos genéticos específicos de PWS ou classes moleculares (19).
Histórico, a PWS é dividida em dois estágios clínicos com falha em prosperar durante a infância representando o primeiro estágio clínico e a hiperfagia com início de obesidade representando o segundo estágio (2). Posteriormente, as fases nutricionais foram descritas para este distúrbio genético relacionado à obesidade e incluem: Fase 0 com diminuição do movimento fetal e retardo do crescimento no útero, seguida da Fase 1 relacionada à hipotonia, falha em se desenvolver com dificuldade de alimentação, Fase 2 começando com ~2 anos de idade quando o ganho de peso é notado pela primeira vez e Fase 3 quando a falta de saciedade é acompanhada pela busca de alimentos e hiperfagia levando à obesidade, se não controlada externamente. A Fase 3 começa por volta dos 6-8 anos de idade (20).
Síndrome de Angelman é caracterizada por um atraso no desenvolvimento muitas vezes não aparente até cerca dos 6 meses de idade e subsequente início de crises muitas vezes difíceis de controlar, tremores, marcha ampla e ataxia com um comportamento feliz característico (3). Existem quatro mecanismos moleculares reconhecidos do AS: de novas deleções maternas do cromossomo 15q11-q13 (70-80%); mutações do gene UBE3A herdado maternalmente (10-20%); disomia paterna 15 (3-5%); e defeitos de impressão (3-5%) dentro da região 15q11-q13 que alteram a expressão do gene UBE3A causal (21).
Indivíduos com AS frequentemente não são notados por profissionais médicos até ~6 meses de idade quando são relatados atrasos no desenvolvimento, em particular atrasos no desenvolvimento motor. Nessa altura, os pais podem reconhecer o comportamento feliz que inclui risos frequentes, sorrisos e excitabilidade. Uma menor necessidade de sono é relatada em >80% dos indivíduos com AS (22). Eles frequentemente desenvolvem convulsões aos 1-3 anos de idade (23). A epilepsia pode ser intratável e tem uma aparência característica no EEG descrita como um aumento do poder delta com uma onda trifásica característica. Indivíduos com AS são descritos como atáxicos em seus movimentos e caminhadas (24, 25). A microcefalia pode se desenvolver até ~2 anos de idade. Os comportamentos estereotipados incluem amor à água e papel enrugado e os indivíduos com SA são caracteristicamente não-verbais e categorizados como severamente incapacitados intelectualmente. Entretanto, é notável que indivíduos com SA não têm habilidades bem capturadas nos testes neuropsicológicos objetivos atualmente disponíveis. Eles têm fortes habilidades na manipulação eletrônica, mas os comportamentos podem ser desafiadores e incluir ansiedade com curtos períodos de atenção.
As pacientes com SA podem apresentar fenótipos variáveis dependendo da classe molecular e como existem abordagens potenciais de tratamento e vigilância para cada um, um fluxograma lógico é necessário para ordenar testes genéticos pelo médico que avalia esses pacientes. O foco do nosso relatório é descrever os achados clínicos e genéticos desses dois distúrbios de impressão genômica e ilustrar as opções de testes genéticos disponíveis no ambiente clínico e a ordem em que os diferentes testes genéticos podem ser obtidos de forma mais produtiva.
Experiência Genética Laboratorial nos Distúrbios de Impressão do Cromossoma 15
Síndrome de Prader-Willi
Para servir como exemplo da importância dos testes de microarranjo SNP de alta resolução, uma grande coorte de 510 participantes com PWS confirmada geneticamente foram recrutados nos EUA e agrupados em três classes moleculares. Eles foram ainda caracterizados como subtipos de deleção 15q11-q13, disomia materna 15 subclasses e defeitos do centro de impressão (16). Nesta maior coorte de PWS relatada, 303 indivíduos foram encontrados com a deleção 15q11-q13 (60% dos casos) composta de 118 indivíduos (38,9%) tendo a maior deleção típica 15q11-q13 tipo I envolvendo os pontos de quebra do cromossomo 15q11-q13 BP1 e BP3 e 165 indivíduos (54.5%) teve a menor deleção típica 15q11-q13 Tipo II envolvendo pontos de quebra BP2 e BP3 com 20 indivíduos tendo uma deleção atípica maior ou menor que a típica 15q11-q13 (6,6%). Em pessoas identificadas como tendo uma deleção do cromossomo 15, é importante considerar se uma translocação balanceada poderia estar presente no pai do probando, pois isso aumenta o risco de recorrência de PWS na descendência do pai. Para a disomia materna 15, 185 indivíduos (36%) apresentaram a disomia uniparental materna 15 (UPD15) com 13 indivíduos (12,5%) com isodisomia total de todo o cromossomo 15 devido a erros na meiose materna II; 60 (57,7%) apresentaram isodisomia segmentar por eventos cruzados na meiose materna I e 31 apresentaram heterodisomia (29,8%), enquanto 81 indivíduos não tiveram a análise do microarranjo SNP e a classificação da disomia materna 15 determinada. Em relação aos defeitos de imprinting PWS, 22 indivíduos (4%) foram encontrados com 13 (76,5%) tendo um status de epimutação sem depleção, quatro indivíduos (23,5%) tiveram uma microdeleção do centro de imprinting enquanto os cinco indivíduos restantes não tiveram um tipo de defeito de imprinting estabelecido. Em um estudo relacionado, uma análise adicional dos defeitos de imprinting na PWS foi realizada por Hartin et al. (8) usando PCR digital de gotículas e seqüenciamento de toda a próxima geração em uma coorte separada de 15 pacientes não relacionados e dois indivíduos ou 13% foram encontrados com um defeito de microdeleção do centro de imprinting. Nos 60 indivíduos com isodisomia segmentar 15 relatados por Butler et al. (16), o tamanho médio total da perda de heterozigossidade (LOH) foi de 25,1 Mb com um intervalo de 5-67,4 Mb e um tamanho médio de 16,4 Mb para LOHs individuais. Trinta e dois indivíduos tinham um segmento LOH, 25 indivíduos tinham dois segmentos e três indivíduos tinham três segmentos. Os locais LOH mais comuns foram a região proximal 15q11-q13 e a região distal 15q26 incluindo as bandas 15q12 e 15q26.1 como mais comumente registradas.
A presença de UPD15 materno e determinação de subclasse específica (isodisomia segmentar ou total) pode impactar o diagnóstico e a vigilância médica como uma segunda condição genética pode estar presente se a mãe for portadora de um alelo genético recessivo localizado na região LOH levando a duas cópias idênticas. Centenas de genes potencialmente causadores de doenças são encontrados no cromossomo 15 e essas doenças devem ser verificadas ou monitoradas de perto naquelas com isodisomia segmentar ou total do cromossomo 15. Um fluxograma de testes genéticos proposto para identificar as diferentes classes moleculares tanto para pacientes com PWS quanto para pacientes com AS pode ser visto no Abstract Gráfico.
Síndrome de Angelman
Quatro classes moleculares reconhecidas foram identificadas no AS que podem ser categorizadas pelo impacto na metilação da região do cromossomo 15. O subtipo mais comum é uma deleção do 15q11 materno.2-q13 região como visto similarmente de origem paterna em PWS e encontrado em ~70% dos indivíduos com AS (21). Entretanto, no AS a eliminação típica de Classe II é mais comum. Esta deleção típica de Classe II menor aproxima-se mais comumente de 5 Mb no tamanho da BP2-BP3 e está presente em 50% dos casos de deleção de AS. As deleções de Classe I são de 5-7 Mb de tamanho e abrangem BP1-BP3 (40% dos casos de deleção). As deleções atípicas podem se estender de BP1 ou BP2-BP4 ou pontos de interrupção mais distantes. Em indivíduos com uma deleção na cópia materna do cromossomo 15, deve-se considerar se há sinais no microarranjo cromossômico mostrando distúrbios que indicam que pode haver uma translocação materna. Isso aumenta o risco de recorrência do AS em futuros descendentes maternos. A disomia paternal uniparental 15 é responsável por 5-7% dos indivíduos com AS. Os defeitos de impressão são responsáveis por 3-5% dos indivíduos com AS e são causados por defeitos no centro de controle de impressão resumidos por Buiting et al. (26). Em indivíduos com defeito no centro de controle do imprint, a marcação epigenética na linha germinal não muda adequadamente de um padrão paterno com expressão silenciosa UBE3A para permitir um padrão materno de expressão no gene UBE3A. Em até 50% dos casos relatados, uma mutação no centro de controle do imprinting pode ser identificada. Casos em mosaico de defeitos no centro de impressão em que uma porcentagem de células não tem expressão na região 15q11.2-q13 é relatada e pode ser mais comum do que se pensava anteriormente (27). O defeito genético final no AS não tem impacto nos resultados dos testes de metilação do DNA, mas é causado por uma mutação no gene UBE3A herdado maternalmente. As mutações neste gene representam 11% dos casos de AS (28). Uma mutação UBE3A pode ser herdada maternalmente e, portanto, é indicado fazer testes direcionados na mãe da paciente para descartar um risco de 50% de recorrência em sua futura descendência. Se a mutação for considerada hereditária, recomendamos que se considere a possibilidade de testar o avô materno da paciente, pois isso poderia ter implicações para os futuros filhos da tia materna.
Discussão
O manejo médico de PWS e AS deve ser dirigido por uma equipe multidisciplinar durante a infância. Ambos os bebês com PWS (mais comumente) e AS podem ter falha em prosperar. Um dietista desempenha um papel importante nos cuidados no início para lidar com o fracasso no desenvolvimento e mais tarde na infância para evitar a obesidade com intervenção dietética com restrição e uso de programas de exercício (que é uma preocupação notada mais comumente para a SPW, mas agora reconhecida no AS em alguns indivíduos). Os geneticistas clínicos, especialistas em ortopedia, médicos de cuidados primários, terapeutas especializados em medicina do trabalho (OT), física (PT) e fala (SLP), especialistas em saúde mental, especialistas em sono, especialistas em saúde mental e endocrinologistas são necessários para abordar as múltiplas questões de saúde na SPV que podem ocorrer. Uma equipe AS inclui geneticistas clínicos, neurologistas, terapeutas especializados em PT, OT e SLP, especialistas em sono, gastroenterologia, medicina física e reabilitação, ortopedia, e especialistas em saúde mental. Para PWS, cuidados médicos apropriados, gerenciamento e aconselhamento são objetivos para controlar o ganho de peso e monitorar e tratar condições comorbitárias, comportamento e problemas psiquiátricos associados. O crescimento e outras deficiências hormonais comuns nesta doença requerem tratamento. O controle rigoroso da dieta com segurança alimentar e um ambiente de rotina controlado com exercício regular são estratégias importantes para controlar a hiperfagia, obesidade e complicações relacionadas necessárias ao longo da vida. AS requer intervenção precoce, incluindo conhecimento de intervenções terapêuticas especializadas, como dispositivos de comunicação aumentativa e assistiva e um programa de fortalecimento de exercícios e atividades de desenvolvimento intensivo para atingir o potencial máximo (por exemplo, SPIDER), tratamento precoce com benzodiazepinas para convulsões e terapia alimentar, como o uso de uma dieta cetogênica. A maximização de todos os aspectos dos cuidados, incluindo os distúrbios do sono e a constipação, influenciam grandemente o controle das convulsões. Um centro especializado familiarizado com as complexidades e aspectos únicos destes distúrbios pode afetar o resultado.
O diagnóstico precoce é vital para garantir uma intervenção precoce tanto para a SPV como para a SA. Para a SPW, um diagnóstico precoce deve ser feito durante a infância para iniciar o tratamento do hormônio de crescimento, controlar as preocupações com a alimentação, obesidade, deficiências hormonais, atrasos no desenvolvimento e problemas comportamentais. O diagnóstico no AS também assegura terapias precoces que impactam os resultados do desenvolvimento, bem como a profilaxia de convulsões, incluindo a preparação com benzodiazepinas apropriadas. Outras intervenções que podem ser benéficas incluem dietas especializadas para indivíduos com SA, como a dieta cetogênica ou terapia de baixo índice glicêmico (LGIT). O diagnóstico precoce também pode reduzir os custos dos cuidados médicos, prevenindo hospitalizações prolongadas relacionadas a problemas de alimentação em indivíduos com SPW e convulsões para crianças com AS.
Identificar a classe molecular de SPW ou AS com testes genéticos avançados, tais como microarrays de SNP de alta resolução, permitirá um diagnóstico mais preciso, levando a melhores indicadores para o prognóstico, e um aconselhamento genético mais preciso dos membros da família. Microarrays SNP de alta resolução, análise FISH, amplificação da sonda de ligadura por metilação múltipla específica (MS-MLPA) e/ou genotipagem do cromossoma 15 são todos úteis na determinação das deleções 15q11-q13. Microarrays SNP de alta resolução podem identificar os subtipos de deleção (típicos e atípicos) nas subclasses PWS e AS, e UPD15 (isodisomia segmentar, e isodisomia total). O subtipo de heterodisomia de UPD e defeitos IC (microdeleção e epimutação) em ambas as subclasses PWS e AS pode requerer um trabalho diagnóstico adicional, conforme ilustrado no Resumo Gráfico. O subtipo ou classes tem impacto no diagnóstico, risco potencial de recorrência para os membros da família, prognóstico e monitoramento de outras condições genéticas e características de alto risco relacionadas com a classe molecular. Por exemplo, características autistas e psicose são mais comuns naqueles com PWS e disomia materna 15 e podem estar relacionadas com as subclasses específicas da UPD15. Aqueles com as maiores deleções de Classe I no AS têm maior probabilidade de desenvolver convulsões difíceis de tratar e microcefalia.
Um fluxograma de testes genéticos incorporando opções de testes que estão disponíveis, incluindo aqueles usados historicamente tanto para a PWS quanto para o AS estão listados no Resumo Gráfico. Os testes para PWS ou AS muitas vezes começam com a metilação do DNA e, se anormal, avança para outros métodos de testes genéticos incluindo microarrays SNP de alta resolução ou ensaios MS-MLPA com base na disponibilidade para os clínicos e famílias em seu ambiente clínico. De preferência, uma matriz de SNP de alta resolução seria encomendada, a qual está pronta e comercialmente disponível no atendimento médico Westernized. O sequenciamento de próxima geração (NGS) do exoma (ou genoma inteiro) também está disponível para os clínicos, mas a PCR digital de gotículas (ddPCR) é atualmente baseada em pesquisa (14). As matrizes de SNP podem identificar classes moleculares específicas na maioria dos pacientes que apresentam características de PWS (cerca de 85% dos casos) ou AS (cerca de 80%), enquanto os pacientes restantes necessitarão de testes adicionais, conforme descrito no Abstract Gráfico. Testes genéticos avançados específicos (por exemplo, ddPCR) podem ser adequadamente sensíveis para quantificar o mosaicismo e podem identificar um diagnóstico em um grande subconjunto de indivíduos com características clínicas mais leves de PWS e AS, mas mais pesquisa é necessária.
Foram encontradas diferenças clínicas iniciais quando comparados aqueles com PWS ou AS com o estado de deleção vs. não deleção (29), incluindo hipopigmentação naqueles com PWS e AS com deleção 15q11-q13 (30). Mais tarde, foram relatados resultados de QI verbal (31) ou psicose (32) mais elevados naqueles com UPD15 materna em comparação com a deleção em indivíduos com SPW. Além disso, Butler et al. (19) relataram menores escores adaptativos e comportamentos mais obsessivos-compulsivos em indivíduos com a deleção 15q11-q13 tipo I em comparação com a UPD15. Zarcone et al. (33) relataram que indivíduos com PWS e a deleção 15q11-q13 tipo I tinham mais compulsões com limpeza pessoal e comportamento compulsivo que era mais difícil de interromper e interferir nas atividades sociais do que aqueles com deleções tipo II ou UPD15. Em um estudo de correlação fenótipo-genótipo na síndrome de Angelman, Moncla et al. (34) relataram aumento da atividade convulsiva naqueles com a deleção de Classe I maior em comparação com a não-eleição. Microcefalia, ataxia, hipotonia e dificuldades de alimentação também são mais prováveis no subtipo de deleção (3). Elas podem ter um comprometimento mais severo da linguagem em particular da linguagem receptiva e traços autistas (21, 35). Indivíduos com SA com UPD paterno podem ter linguagem receptiva melhorada, habilidades motoras melhoradas e uma prevalência reduzida de convulsões. Indivíduos com mosaico também podem ter um fenótipo mais brando, incluindo melhoria da linguagem, funcionamento adaptativo e menos convulsões (36).
Exoma de última geração ou sequenciação de genes inteiros também podem ter um lugar nas avaliações genéticas em PWS ou AS, particularmente nos indivíduos que apresentam achados incomuns ou diagnóstico tardio (por exemplo, UPD15 isodisomia segmentar ou total) e nos casos em que o DNA parental não está disponível (8). Para abordar o uso e o tipo de testes genéticos para a PWS e AS, foi desenvolvido um novo fluxograma de testes genéticos para o clínico, conforme descrito e ilustrado no Abstract Gráfico. Este fluxograma pode ajudar a ordenar testes genéticos baseados na apresentação clínica para determinar o diagnóstico, manejo e tratamento apropriados e fornecer as informações mais precisas de aconselhamento genético para outros membros da família. Sugerimos o uso deste algoritmo para completar definitivamente o trabalho de diagnóstico tanto para PWS como para AS. Argumentamos que o diagnóstico é incompleto sem o conhecimento do subtipo genético específico do paciente para orientar o aconselhamento, a orientação antecipatória, o manejo e as prováveis opções terapêuticas. A determinação da classe molecular é importante para o cuidado e tratamento médico e útil para o clínico envolvido no aconselhamento genético de familiares para PWS ou AS.
Contribuições do Autor
MB e JD contribuíram para a redação do manuscrito, revisão de literatura, contribuíram com sua experiência e editaram o manuscrito.
Funding
Reconhecemos o número de bolsa HD02528 do Instituto Nacional de Saúde Infantil e Desenvolvimento Humano.
Conflito de Interesses
Os autores declaram que a pesquisa foi realizada na ausência de qualquer relação comercial ou financeira que pudesse ser interpretada como um potencial conflito de interesses.
Conhecimento
Conhecemos Grace Graham pela preparação especializada do manuscrito.
1. Bittel DC, Mordomo MG. Síndrome de Prader-Willi: genética clínica, citogenética e biologia molecular. Especialista Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Gestão da Síndrome de Prader-Willi. Nova York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Aspectos clínicos e genéticos da síndrome de Angelman. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Síndrome de Prader-Willi. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Síndrome de Prader-Willi: uma revisão dos achados clínicos, genéticos e endócrinos. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Gene único e causas sindrômicas de obesidade: exemplos ilustrativos. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting sugerido por heterodisomia materna na síndrome de Prader-Willi não-deleção. Natureza. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Análise do centro de impressão da síndrome de Prader-Willi usando PCR digital de gotículas e sequenciamento de toda a próxima geração. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Três irmãos com síndrome de Prader-Willi, causada por microdeleções do centro de impressão e revisão. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Síndrome de Prader-Willi e deleções submicroscópicas atípicas 15q11-q13 com ou sem defeitos de impressão. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Síndrome de Prader-Willi e síndrome de Schaaf-Yang: doenças do desenvolvimento neurológico que se intersectam no gene MAGEL2. Doenças. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Fenótipo Prader-Willi causado por deficiência paterna para o HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Fenótipo Prader-Willi-Like causado por uma microdeleção atípica 15q11.2. Genes (Basiléia). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deleções do cromossoma 15 como causa da síndrome de Prader-Willi. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Mordomo MG, Palmer CG. Origem dos pais da eliminação do cromossoma 15 na síndrome de Prader-Willi. Lanceta. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Classificação genética molecular na síndrome de Prader-Willi: um estudo de coorte multissítios. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, síndrome de Thompson T. Prader-Willi: achados clínicos e genéticos. Endocrinologista. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Mordomo MG. Síndrome de Prader-Willi: entendimento atual da causa e diagnóstico. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Diferenças comportamentais entre sujeitos com síndrome de Prader-Willi e deleção tipo I ou tipo II e disomia materna. Pediatria. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Fases nutricionais na síndrome de Prader-Willi. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Fenótipos distintos distinguem as classes moleculares da síndrome de Angelman. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a casecontrolled study. Psiquiatria de Frente. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Cartão genético de utilidade clínica para: Síndrome de Angleman. Genet Eur J Hum. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Comportamento e manifestações neuropsiquiátricas na síndrome de Angelman. Tratamento neuropsiquiátrico. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. Uma visão geral dos problemas de saúde e desenvolvimento em uma grande coorte clínica de crianças com síndrome de Angelman. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelman syndrome – insights into a rare neurogenetic disorder. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Síndrome de Angelman atípico devido a um defeito de impressão do mosaico: relatos de casos e revisão da literatura. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Síndrome de Angelman. Neuroterapêutica. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Pesquisa clínica e citogênica de 39 indivíduos com síndrome de Prader-Labhart-Willi. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Mordomo MG. Hipopigmentação: uma característica comum da síndrome de Prader-Labhart-Willi. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Telhado E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Características intelectuais da síndrome de Prader-Willi: comparação de subtipos genéticos. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Doença psicótica em pessoas com síndrome de Prader-Willi devido à disomia uniparental materna do cromossoma 15. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. A relação entre comportamento compulsivo e realização acadêmica através dos três subtipos genéticos da síndrome de Prader-Willi. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Correlação fenótipo-genótipo em 20 pacientes com síndrome de Angelman de eliminação e 20 pacientes sem síndrome de Angelman de não eliminação. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, alemão JR, Shaw CA, Bird LM, Kimonis V, et al. Identificação de novas supressões de 15q11q13 na síndrome de Angelman por array-CGH: caracterização molecular e correlações genótipo-fenótipo. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Linguagem expressiva preservada como determinante fenotípico da síndrome de Angelman do mosaico. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar