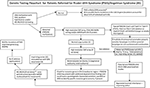
Graphical Abstract. Geneettisen testauksen vuokaavio Prader-Willin oireyhtymän (PWS)/Angelmanin oireyhtymän (AS) vuoksi lähetetyille potilaille. *Rutiininomaisilla kromosomitutkimuksilla suljetaan pois Chr15-translokaatiot tai -inversiot; harkitaan muita lihavuuteen liittyviä geneettisiä häiriöitä; saattaa vaatia fragiili X -oireyhtymän DNA-seulontaa FMR1-geenin toiston laajenemisen varalta tai pitkälle menevää geneettistä testausta seuraavan sukupolven sekvensoinnilla (NGS) FMR1:n tai muiden kandidaattigeenien varianttien varalta käyttäen koko-eksomisekvensointia (Whole Exome Sequencing, WES) tai kokonaisen geeniperimän sekvensointia (Whole Genome Sequencing, WGS); esim. monogeeniset liikalihavuuteen liittyvät syyt). **Voidaan käyttää muiden Chr15-imprinting-geenien metylaatiotilanteen tarkistamiseen; ddPCR:ää, droplet digital PCR:ää voidaan käyttää mosaiikin seulontaan.
Esittely
Kromosomi 15:n imprinting-häiriöihin kuuluvat Prader-Willin (PWS) ja Angelmanin (AS) oireyhtymät (1-6) sekä kromosomi 15q:n duplikaatiot. PWS:n tai AS:n diagnoosi riippuu alkuperäisestä vanhemmasta ja siitä, rajoittuuko ilmentyminen poikkeavasti äidin vai isän imprintoituneisiin geeneihin. Duplikaatio 15q johtuu ylimääräisestä kopiosta äidistä peräisin olevalla 15q11.2-q13-alueella, mikä voi johtaa kouristuskohtauksiin, kognitiivisiin ja käyttäytymiseen liittyviin ongelmiin, mukaan lukien autismikirjon häiriö (ASD), mutta ei PWS- tai AS-fenotyyppiin. PWS johtuu kromosomissa 15q11-q13 sijaitsevien, äidin kautta painautuneiden ja isän kautta ilmentyneiden geenien katoamisesta, kun taas AS johtuu tällä alueella sijaitsevien, äidin kautta painautuneiden ja äidin kautta ilmentyneiden geenien katoamisesta, mikä vaikuttaa erityisesti UBE3A-geeniin. Koska vastuussa olevat geenit ovat imprintoituneita, sekä geneettiset että epigeneettiset virheet voivat olla aiheuttajia.
Vuonna 1989 todettiin, että henkilöillä, joilla oli sekä PWS että ei-deleetio-status, oli äidiltä peräisin oleva äidin disomia 15 tai molemmat 15:t, kun käytettiin proksimaaliselta 15q11-q13-alueelta peräisin olevia polymorfisia DNA-markkereita (7). Myöhemmin 1990-luvun puolivälissä kehitettyjen fluoresoivien in situ -hybridisaation (FISH) DNA-koettimien avulla tunnistettiin 15q11-q13-alueen deleetioita sekä PWS:ssä että AS:ssä. Tänä aikana kehitettiin metylaatio-DNA-testaus, ja PWS:ssä ja AS:ssä havaittiin epänormaali metylaatiokuvio. Metylaatio-DNA-testaus on ~99 % tarkka PWS-diagnoosin tunnistamisessa, mutta sillä ei voida tunnistaa yksittäistä PWS-molekyyliluokkaa (2). AS:n osalta metylaatio-DNA-testi tunnistaa ~80 % yksilöistä, mutta ei myöskään erota molekyyliluokkia toisistaan eikä havaitse AS:n aiheuttavaa mutaatiota UBE3A-geenissä.
Mikrosirutekniikka kehitettiin 2000-luvun alussa tai puolivälissä, ja se edisti diagnostiikan tuottoa. Nyt uusissa SNP-mikrosarjoissa on yli kaksi miljoonaa DNA-koetinta, ja ne ovat käyttökelpoisia deletioalatyyppien ja UPD15-alaluokkien havaitsemisessa. Muu teknologia, kuten droplet digital PCR (ddPCR), määrittelee kopioluvun kromosomi 15:n DNA-koettimien avulla, ja sillä voidaan diagnosoida PWS:n tai AS:n geenivirheitä (8). Lisäksi SNP-mikrosirujen avulla voidaan tunnistaa LOH:t, jotka on määritelty kooltaan >8 Mb:n suuruisiksi, ja jos niitä esiintyy kromosomissa 15, ne tukevat äidin disomia 15:n tai isän disomia 15:n diagnoosia PWS:n tai AS:n epänormaalin DNA-metylaatiokuvion esiintyessä. Imprinting-defektin vahvistaminen saattaa vaatia SNP-mikrosiruja pienten mikrodeleetioiden tunnistamiseksi, mutta myös vanhempien DNA-näytteitä genotyypin määrittämiseksi, jotta voidaan tunnistaa kromosomi 15:n normaali (biparentaalinen) periytyminen, joka tukee epimutation imprinting-defektin esiintymistä PWS:ssä tai AS:ssä ja vaikuttaa siten uusiutumisriskiin. IC-mikrodeleetion erottaminen ei-deleetionaalisesta epimutoituneesta statuksesta on kliinisesti tärkeää perheille, sillä jos vanhemmalla todetaan IC-mikrodeleetio, on 50 prosentin uusiutumisriski lisälapsille (9).
15q11-q13-alueella on yli kymmenkunta geeniä ja transkriptiota, joilla näyttäisi olevan merkitystä PWS:n ja/tai AS:n synnyssä. Geenejä ja transkripteja, jotka sisältyvät proksimaalisesta 15q11.2-murtumakohdasta BP1 ja distaalisesta 15q13-murtumakohdasta BP3 alkavalle alueelle, ovat TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, ei-koodaavat RNA:t (SNORDit), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 ja HERC2. Painetut MRKN3-, MAGEL2-, NDN-, NIPAP1- ja SNURF-SNRPN-geenit ilmentyvät isän puolelta, ja häiriintyneinä ne voivat aiheuttaa PWS-oireita. Esimerkiksi MAGEL2-geenin mutaatiot voivat aiheuttaa vastasyntyneen hypotoniaa, kehitysviivästymää, artrogrypoosia, autistisia piirteitä, huonoa imukykyä ja lihavuutta . On myös raportoitu potilaita, joilla on PWS:n piirteitä ei-koodaavan SNORD116-transkriptin pienten deleetioiden (12) ja muiden samankaltaisten deleetioiden seurauksena (10, 13).
Keskityimme tässä raportissa AS:ään ja PWS:ään, koska molemmat oireyhtymät havaitaan DNA:n metylaatiotesteillä, jotka mahdollistavat aktiivisen vanhemman geenin alleelin määrittämisen ja lopullisen diagnoosin PWS:ää sairastavilla henkilöillä ja suurimmalla osalla AS:ää sairastavista (2). DNA-metylaatiotesteillä ei kuitenkaan voida tunnistaa molekyyliluokkaa kummassakaan oireyhtymässä. Korkearesoluutioinen kromosomianalyysi kehitettiin ja sitä käytettiin 1980-luvun alussa, ja siitä tuli standardilaboratorion geneettiseen testiin perustuva testi, jolla arvioitiin kromosomin 15q11-q13 deleetio, joka tunnistettiin tuolloin suurimmalla osalla PWS-potilaista (14), ja myöhemmin AS:n osalta. Vuonna 1983 raportoitiin 15q11-q13-deleetion isänpuoleisesta alkuperästä (15), ja sen todettiin olevan de novo, mutta 15q11-q13-deleetion kokoa tai tyyppiä (tyypillinen vs. epätyypillinen) ei voitu määrittää. Tarkka ja varhainen diagnoosi, johon liittyy molekyyliluokan tunnistaminen, on olennaisen tärkeää paitsi kliinisen diagnoosin vahvistamiseksi myös geneettisen neuvonnan, hoidon ja hoidon tiedottamisen ja odotusten ohjaamisen kannalta. Meneillään olevien kliinisten tutkimusten myötä molekulaarisen etiologian parempi ymmärtäminen voi vaikuttaa potilaan osallistumismahdollisuuksiin. Lisäksi lähestyviin tutkimuksiin kuuluu antisense-oligonukleotideja kromosomin 15 vaiennetun isänpuoleisen kopion aktivoimiseksi uudelleen henkilöillä, joilla on AS.
PWS ja AS ovat monimutkaisia harvinaisia neurologisia kehityshäiriöitä, jotka johtuvat virheistä genomisessa imprintingissä. PWS tunnustetaan yleisimmäksi geneettiseksi syyksi henkeä uhkaavaan liikalihavuuteen, jos se jätetään hallitsematta (2, 4, 6). Tunnettuja PWS:n molekyyliluokkia on kolme, mukaan lukien isänpuoleinen 15q11-q13-deleetio, joka on kooltaan noin 5-6 Mb (60 % tapauksista), ja äidinpuoleinen 15-disomia (UPD15), jossa molemmat 15-kromosomit periytyvät äidiltä (36 %) ja joka on peräisin trisomia 15:stä, jossa isänpuoleinen 15-kromosomi katoaa raskauden alkuvaiheessa, mikä johtaa siihen, että äidiltä periytyy kaksi kromosomia 15:stä (16). Kolmas luokka on leimautumiskeskuksen vika. Jos isän alleelissa on mikrodeleetio tai epimutaatio imprinting-keskuksessa (IC), joka kontrolloi kromosomissa 15 olevien valittujen imprinting-geenien ilmentymistilaa, syntyy PWS. Tätä imprinting-virhettä esiintyy 4 prosentilla henkilöistä, joilla on PWS (8, 16). Useimmat PWS-tapaukset ovat sporadisia, ja etnisten ryhmien ja sukupuolen välinen osuus on suunnilleen sama. PWS:n arvioitu esiintyvyys on yksi 10 000:sta yhteen 30 000:sta (2). Maailmanlaajuisesti PWS:ää sairastavia henkilöitä arvioidaan olevan noin 400 000, joista noin 20 000 asuu Yhdysvalloissa (2, 17).
PWS:lle on ominaista lapsuusiän hypotonia, heikko imemisrefleksi, johon liittyy ruokailuvaikeuksia, lyhytkasvuisuus ja pienet kädet ja jalat, hormonipuutoksista johtuva hypogonadismi, lievä älyllinen kehitysvammaisuus, käytöshäiriöt ja hyperfagia, joka alkaa usein 6-8 vuoden iässä ja joka jatkuu aikuisikään asti ja joka voi johtaa lihavuuteen, jos ympäristökontrollit eivät ole kunnossa. Imeväisikäisenä nähdään tyypillisiä kraniofaktisia piirteitä, kuten kapea bifrontaalinen läpimitta, karsastus, pieni ylöspäin kääntynyt nenä ja ohut ylähuuli, alaspäin kääntyneet suun kulmat, tahmea sylki ja kiilteen hypoplasia (2, 4, 6, 18). Kognitio on yleensä heikentynyt perhetaustan perusteella, ja lapsuudessa alkaviin käytösongelmiin kuuluvat itsensä vahingoittaminen (ihon repiminen), purkaukset, itsepäisyys ja kiukkukohtaukset, ja psykiatriset ongelmat ilmenevät tänä aikana tai myöhemmin nuoruusiässä tai nuoressa aikuisuudessa (2). Käyttäytymisongelmiin kuuluvat ahdistuneisuus, mielialahäiriöt, psykoosi ja autismi, jotka voivat korreloida tiettyjen PWS:n geneettisten alatyyppien tai molekyyliluokkien kanssa (19).
Historiallisesti PWS on jaettu kahteen kliiniseen vaiheeseen, joissa imeväisiässä tapahtuva menestymättömyys edustaa ensimmäistä kliinistä vaihetta ja hyperfagia, johon liittyy liikalihavuuden puhkeaminen, toista vaihetta (2). Myöhemmin tälle lihavuuteen liittyvälle geneettiselle sairaudelle on kuvattu ravitsemuksellisia vaiheita, joihin kuuluvat: Vaihe 0, jossa sikiön liikkeet vähenevät ja kasvu hidastuu kohdussa, jota seuraa vaihe 1, joka liittyy hypotoniaan, menestymättömyyteen ja ruokailuvaikeuksiin, vaihe 2, joka alkaa noin kahden vuoden iässä, kun painonnousua havaitaan ensimmäisen kerran, ja vaihe 3, jolloin kylläisyyden puutteeseen liittyy ruoan etsimistä ja hyperfagiaa, joka johtaa liikalihavuuteen, jos sitä ei valvota ulkoisesti. Vaihe 3 alkaa noin 6-8 vuoden iässä (20).
Angelmanin oireyhtymälle on ominaista kehitysviive, joka ilmenee usein vasta noin 6 kuukauden iässä, ja sen jälkeen alkavat usein vaikeasti hallittavissa olevat kouristuskohtaukset, vapina, leveäpohjainen kävely ja ataksia, joihin liittyy tyypillinen iloinen käytös (3). AS:n molekyylimekanismeja on tunnistettu neljä: kromosomin 15q11-q13 de novo -deleetio äidissä (70-80 %), äidin perimän UBE3A-geenin mutaatiot (10-20 %), isän disomia 15 (3-5 %) ja 15q11-q13-alueella esiintyvät jälkipainautumisvauriot (3-5 %), jotka muuttavat aiheuttavan UBE3A-geenin ilmentymistä (21).
Asiantuntijat huomaavat AS:ää sairastavat henkilöt usein vasta ~6 kuukauden iässä, jolloin kehitysviiveet, erityisesti viivästynyt motorinen kehitys, tulevat ilmi. Tähän mennessä vanhemmat saattavat tunnistaa iloisen käytöksen, johon kuuluu usein naurua, hymyilyä ja innostuneisuutta. Vähentyneen unentarpeen raportoidaan olevan >80 %:lla henkilöistä, joilla on AS (22). Heille kehittyy usein kohtauksia 1-3 vuoden iässä (23). Epilepsia voi olla vaikeahoitoinen, ja sillä on EEG:ssä tyypillinen ulkonäkö, jota kuvataan lisääntyneenä delta-tehona, jossa on tyypillinen trifaattinen aalto. AS:ää sairastavien henkilöiden liikkeitä ja kävelyä kuvataan ataksisiksi (24, 25). Mikrokefalia voi kehittyä noin 2 vuoden iässä. Stereotyyppiseen käyttäytymiseen kuuluu rakkaus veteen ja rypistyneeseen paperiin, ja AS:ää sairastaville henkilöille on tyypillistä, että he eivät puhu ja heidät luokitellaan vaikeasti kehitysvammaisiksi. On kuitenkin huomattava, että AS-tautia sairastavilla henkilöillä on taitoja, joita nykyisin saatavilla olevat objektiiviset neuropsykologiset testit eivät pysty hyvin kuvaamaan. Heillä on vahvat kyvyt elektroniikan manipuloinnissa, mutta käyttäytyminen voi olla haastavaa, ja siihen voi kuulua ahdistuneisuutta ja lyhyttä keskittymiskykyä.
Koska PWS- tai AS-potilailla voi esiintyä vaihtelevia fenotyyppejä molekyyliluokasta riippuen ja koska kummallekin on olemassa potentiaalisia hoito- ja seurantamahdollisuuksia, tarvitaan looginen vuokaavio geneettisten testien tilaamiseksi näitä potilaita arvioivalle kliinikolle. Kertomuksessamme keskitymme kuvaamaan näiden kahden genomipainatushäiriön kliinisiä ja geneettisiä löydöksiä sekä havainnollistamaan kliinisessä ympäristössä käytettävissä olevia geenitutkimusvaihtoehtoja ja järjestystä, jossa eri geenitutkimukset voidaan hankkia tuottavimmin.
Laboratoriogenetiikan kokemuksia kromosomi 15:n impressing-häiriöistä
Prader-Willin oireyhtymä
Esimerkiksi korkearesoluutioisten SNP-mikrosirutestien merkityksestä rekrytoitiin Yhdysvalloissa 510:stä geneettisesti vahvistettua PWS-tautia sairastavasta osallistujasta koostuva laaja monipaikkainen kohortti, joka ryhmiteltiin kolmeen molekulaariseen luokkaan. Niitä luonnehdittiin edelleen 15q11-q13-deleetioiden alatyyppeihin, äidin disomia 15 -alaluokkiin ja imprinting-keskuksen vikoihin (16). Tässä suurimmassa raportoidussa PWS-kohortissa 303 henkilöllä todettiin 15q11-q13-deleetio (60 % tapauksista), joka koostui 118 henkilöstä (38,9 %), joilla oli suurempi tyypillinen 15q11-q13-tyypin I deletio, johon liittyy kromosomin 15q11-q13 murtumispisteet BP1 ja BP3, ja 165 henkilöstä (54.5 %:lla) oli pienempi tyypillinen tyypillinen 15q11-q13 tyyppi II -deleetio, joka liittyy murtumispisteisiin BP2 ja BP3, ja 20 henkilöllä oli epätyypillinen deletio, joka on suurempi tai pienempi kuin tyypillinen 15q11-q13-deleetio (6,6 %). Henkilöillä, joilla on todettu kromosomi 15:n deleetio, on tärkeää harkita, voisiko koehenkilön isällä olla tasapainoinen translokaatio, koska se lisää PWS:n uusiutumisriskiä isän jälkeläisillä. Äidin disomia 15:n osalta 185 henkilöllä (36 %) oli äidin uniparentaalinen disomia 15 (UPD15), ja 13 henkilöllä (12,5 %) oli koko kromosomi 15:n totaalinen isodisomia, joka johtui virheistä äidin meioosi II:ssa; 60:llä (57,7 %) oli segmentaalinen isodisomia, joka johtui ristikkäistapahtumista äidin meioosi I:ssä, ja 31:llä (29,8 %) havaittiin heterodisomia. 81:llä henkilöllä ei suoritettu SNP-mittasarja-analyysiä, eikä määritetty luokittelua äidin kromosoma 15:een. PWS:n imprintointivirheitä löytyi 22 yksilöltä (4 %), joista 13:lla (76,5 %) oli epimutaatio, joka ei ollut deletoitunut, neljällä yksilöllä (23,5 %) oli imprintointikeskuksen mikrodeleetio, kun taas lopuilla viidellä yksilöllä ei ollut määritetty imprintointivirheen tyyppiä. Aiheeseen liittyvässä tutkimuksessa Hartin ja muut (8) tekivät lisäanalyysin PWS:n imprintointivirheistä käyttämällä digitaalista PCR:ää (droplet digital PCR) ja seuraavan sukupolven koko eksomin sekvensointia erillisessä PWS-kohortissa, joka koostui 15:stä sukuun liittymättömästä potilaasta, ja kahdella henkilöllä eli 13 %:lla todettiin imprintointikeskuksen mikrodeleetion vika. Butlerin ja muiden (16) raportoimissa 60 yksilössä, joilla oli segmentaalinen isodisomia 15, heterotsygotian menetyksen (LOH) kokonaiskoko oli keskimäärin 25,1 Mb, vaihteluväli 5-67,4 Mb ja yksittäisten LOH:ien keskimääräinen koko 16,4 Mb. Kolmellakymmenelläkahdella yksilöllä oli yksi LOH-segmentti, 25 yksilöllä oli kaksi segmenttiä ja kolmella yksilöllä oli kolme segmenttiä. Yleisimmät LOH-kohdat olivat proksimaalinen 15q11-q13-alue ja distaalinen 15q26-alue, mukaan lukien 15q12- ja 15q26.1-kaistat yleisimmin kirjattuina.
Äidin UPD15:n esiintyminen ja spesifisen alaluokan (segmentaalinen tai totaalinen isodisomia) määrittäminen voi vaikuttaa diagnoosiin ja sairaanhoidon seurantaan, sillä toinen geneettinen sairaus voi esiintyä, jos äiti on sellaisen resessiivisesti periytyvää geenialleelia kantava, joka sijaitsee LOH-alueella, mikä johtaa kahteen identtiseen kopioon. Kromosomissa 15 on satoja mahdollisesti tauteja aiheuttavia geenejä, ja näitä tauteja olisi tarkistettava tai seurattava tiiviisti niillä, joilla on kromosomin 15 segmentaalinen tai totaalinen isodisomia. Ehdotettu geneettisen testauksen vuokaavio eri molekyyliluokkien tunnistamiseksi sekä PWS- että AS-potilaille on nähtävissä graafisessa tiivistelmässä.
Angelmanin oireyhtymä
AS:ssä on tunnistettu neljä tunnistettua molekyyliluokkaa, jotka voidaan luokitella kromosomi 15:n alueen metylaatioon kohdistuvan vaikutuksen perusteella. Yleisin alatyyppi on äidin 15q11:n deleetio.2-q13-alueella, kuten vastaavalla tavalla on havaittu isältä peräisin olevassa PWS:ssä, ja sitä esiintyy ~70 prosentilla AS:ää sairastavista henkilöistä (21). AS:ssä tyypillinen luokan II deleetio on kuitenkin yleisempi. Tämä tyypillinen pienempi luokan II deletio on tavallisimmin noin 5 Mb:n kokoinen BP2-BP3:n alueelta, ja sitä esiintyy 50 %:lla deleetio-AS-tapauksista. Luokan I deleetioiden koko on 5-7 Mb, ja ne kattavat BP1-BP3:n (40 % deleetiotapauksista). Epätyypilliset deleetiot voivat ulottua BP1:stä tai BP2-BP4:stä tai kauempana sijaitsevista taitekohdista. Henkilöillä, joilla on deletio kromosomi 15:n äidin kopiossa, on harkittava, onko kromosomimikroskooppitutkimuksessa merkkejä häiriöistä, jotka viittaavat siihen, että kyseessä voi olla äidin translokaatio. Tämä lisää AS:n uusiutumisriskiä äidin tulevissa jälkeläisissä. Uniparentaalinen isänpuoleinen 15-disomia on 5-7 prosentilla henkilöistä, joilla on AS. Jälkiintymishäiriöitä on 3-5 %:lla AS-tautia sairastavista henkilöistä, ja ne johtuvat Buitingin ym. yhteenvedon (26) mukaisista vioista jälkiintymiskontrollikeskuksessa. Henkilöillä, joilla on vika imprintin ohjauskeskuksessa, epigeneettinen merkintä sukusoluissa ei onnistu kunnolla siirtymään isänpuoleisesta mallista, jossa UBE3A-geenin ilmentyminen on vaiennettu, jotta UBE3A-geenin ilmentyminen voisi tapahtua äidinpuoleisessa mallissa. Jopa 50 prosentissa raportoiduista tapauksista voidaan tunnistaa mutaatio imprinting-kontrollikeskuksessa. On raportoitu mosaiikkimaisia tapauksia, joissa imprinting-keskuksen vika on sellainen, että osasta soluista puuttuu 15q11.2-q13-alueen ilmentyminen, ja ne saattavat olla yleisempiä kuin aiemmin on luultu (27). AS:n viimeinen geenivirhe ei vaikuta DNA-metylaatiotestien tuloksiin, mutta sen aiheuttaa mutaatio äidin perimässä UBE3A-geenissä. Tämän geenin mutaatiot aiheuttavat 11 prosenttia AS-tapauksista (28). UBE3A-mutaatio voi periytyä äidin kautta, ja siksi on aiheellista tehdä kohdennettu testaus potilaan äidille, jotta voidaan sulkea pois 50 prosentin uusiutumisriski hänen tulevissa jälkeläisissään. Jos mutaation katsotaan periytyvän, suosittelemme harkitsemaan potilaan äidin isoisän testaamista, koska sillä voi olla vaikutuksia äidin tädin tuleviin lapsiin.
Keskustelu
PWS:n ja AS:n lääketieteellistä hoitoa olisi ohjattava moniammatillisen tiimin toimesta imeväisikäisenä. Sekä PWS:ää (yleisemmin) että AS:ää sairastavilla lapsilla voi esiintyä menestymishäiriöitä. Ravitsemusterapeutilla on tärkeä rooli hoidossa aluksi menestymishäiriön korjaamiseksi ja myöhemmin lapsuudessa lihavuuden välttämiseksi ruokavalion rajoittamisella ja liikuntaohjelmien käyttämisellä (mikä on huolenaihe, joka on todettu yleisemmin PWS:n yhteydessä, mutta joka on nyt tunnistettu AS:n yhteydessä joillakin henkilöillä). Kliinisiä geneetikkoja, ortopedian erikoislääkäreitä, perusterveydenhuollon lääkäreitä, erikoistuneita toimintaterapeutteja (OT), fysioterapeutteja (PT) ja puheterapeutteja (SLP), mielenterveysasiantuntijoita, uniasiantuntijoita, mielenterveysasiantuntijoita ja endokrinologeja tarvitaan, jotta voidaan puuttua moninaisiin terveysongelmiin, joita PWS:ssä voi esiintyä. AS-tiimiin kuuluu kliinisiä geneetikkoja, neurologeja, erikoistuneita terapeutteja PT-, OT- ja SLP-palveluihin, uniasiantuntijoita, gastroenterologeja, fysiatrian ja kuntoutuksen asiantuntijoita, ortopedeja ja mielenterveyden asiantuntijoita. PWS:n kohdalla asianmukainen lääketieteellinen hoito, ohjaus ja neuvonta ovat tavoitteita painonnousun hallitsemiseksi ja siihen liittyvien liitännäissairauksien, käyttäytymisen ja psykiatristen ongelmien seuraamiseksi ja hoitamiseksi. Kasvuhormonin ja muiden hormonien puutokset, jotka ovat yleisiä tässä häiriössä, vaativat hoitoa. Ruokavalion tiukka valvonta ja ruokaturva sekä hallittu rutiiniympäristö, johon kuuluu säännöllistä liikuntaa, ovat tärkeitä strategioita hyperfagian, liikalihavuuden ja niihin liittyvien komplikaatioiden hallitsemiseksi, joita tarvitaan koko elämän ajan. AS vaatii varhaista puuttumista, mukaan luettuna tietämys erityisistä terapeuttisista toimenpiteistä, kuten kommunikaatiota tukevista apuvälineistä ja apuvälineistä sekä intensiivisistä kehitysharjoituksista ja toiminnoista koostuvasta vahvistavasta ohjelmasta maksimaalisten mahdollisuuksien saavuttamiseksi (esim. SPIDER), varhaista hoitoa bentsodiatsepiineillä kohtausten vuoksi ja ruokavaliohoitoa, kuten ketogeenisen ruokavalion käyttöä. Kaikkien hoidon osa-alueiden maksimointi, mukaan lukien unihäiriöt ja ummetus, vaikuttavat suuresti kohtausten hallintaan. Erikoistunut keskus, joka tuntee näiden häiriöiden monimutkaisuudet ja ainutlaatuiset näkökohdat, voi vaikuttaa lopputulokseen.
Varhainen diagnoosi on elintärkeää sekä PWS:n että AS:n varhaisen puuttumisen varmistamiseksi. PWS:n osalta varhainen diagnoosi olisi tehtävä imeväisiässä, jotta voidaan aloittaa kasvuhormonihoito, hoitaa ruokintaongelmia, liikalihavuutta, hormonipuutoksia, kehitysviivästymiä ja käyttäytymisongelmia. AS:n diagnoosi takaa myös varhaisen hoidon, joka vaikuttaa kehitystuloksiin, sekä kouristuskohtausten ennaltaehkäisyn, mukaan lukien asianmukaisten bentsodiatsepiinien käyttö. Muita toimenpiteitä, jotka voivat osoittautua hyödyllisiksi, ovat AS-tautia sairastaville tarkoitetut erityisruokavaliot, kuten ketogeeninen ruokavalio tai matalan glykeemisen indeksin hoito (LGIT). Varhainen diagnoosi voi myös alentaa sairaanhoidon kustannuksia ehkäisemällä PWS-tautia sairastavien henkilöiden ruokintaongelmiin ja AS-tautia sairastavien lasten kouristuskohtauksiin liittyviä pitkiä sairaalahoitojaksoja.
PWS- tai AS-molekyyliluokan tunnistaminen edistyneillä geneettisillä testeillä, kuten korkearesoluutioisilla SNP-mikroskooppitutkimuksilla, mahdollistaa tarkemman diagnoosin, mikä johtaa parempiin ennusteindikaattoreihin ja tarkempaan geneettiseen perinnöllisyysneuvontaan perheenjäsenten osalta. Korkearesoluutioiset SNP-mikrosarjat, FISH-analyysi, metylaatiospesifinen moninkertainen ligaatioanturin amplifikaatio (MS-MLPA) ja/tai kromosomi 15:n genotyypin määritys ovat kaikki hyödyllisiä 15q11-q13-deleetioiden määrittämisessä. Korkean resoluution SNP-mikrosirujen avulla voidaan tunnistaa deletioiden alatyypit (tyypilliset ja epätyypilliset) sekä PWS:ssä että AS:ssä sekä UPD15:n alaluokat (segmentaalinen isodisomia ja täydellinen isodisomia). UPD:n heterodisomian alatyyppi ja IC-virheet (mikrodeleetio ja epimutaatio) sekä PWS:ssä että AS:ssä saattavat vaatia lisädiagnostiikkaa, kuten graafisessa yhteenvedossa on esitetty. Alatyyppi tai -luokat vaikuttavat diagnoosiin, perheenjäsenten mahdolliseen uusiutumisriskiin, ennusteeseen ja muiden geneettisten sairauksien ja molekyyliluokkaan liittyvien korkean riskin piirteiden seurantaan. Esimerkiksi autistiset piirteet ja psykoosi ovat yleisempiä henkilöillä, joilla on PWS ja äidin disomia 15, ja ne voivat liittyä tiettyihin UPD15-alaluokkiin. Niillä, joilla on AS:n suuremmat I-luokan deleetiot, esiintyy todennäköisemmin vaikeasti hoidettavia kouristuksia ja mikrokefaliaa.
Geettisen testauksen vuokaavio, joka sisältää saatavilla olevat testausvaihtoehdot, mukaan lukien ne, joita on historiallisesti käytetty sekä PWS:n että AS:n kohdalla, on lueteltu graafisessa tiivistelmässä. PWS:n tai AS:n testaus aloitetaan usein DNA-metylaatiolla, ja jos se on epänormaalia, siirrytään muihin geneettisiin testausmenetelmiin, kuten korkearesoluutioisiin SNP-mikrosarjoihin tai MS-MLPA-määrityksiin, sen mukaan, miten kliiniset lääkärit ja perheet voivat käyttää niitä kliinisessä ympäristössä. Mieluiten tilattaisiin korkearesoluutioinen SNP-mikrosarja, joka on helposti ja kaupallisesti saatavilla länsimaisessa terveydenhuollossa. Eksomin (tai koko genomin) seuraavan sukupolven sekvensointi (NGS) on myös kliinikoiden saatavilla, mutta pisaran digitaalinen PCR (ddPCR) on tällä hetkellä tutkimuspohjainen (14). SNP-määrityksillä voidaan tunnistaa tietyt molekyyliluokat suurimmalla osalla potilaista, joilla on PWS:n (noin 85 % tapauksista) tai AS:n (noin 80 %) piirteitä, kun taas loput potilaat tarvitsevat lisätestejä, kuten graafisessa yhteenvedossa on kuvattu. Erityiset kehittyneet geneettiset testit (esim. ddPCR) voivat olla riittävän herkkiä mosaikismin kvantifioimiseksi, ja ne voivat tunnistaa diagnoosin suurella osajoukolla henkilöitä, joilla on lievempiä PWS:n ja AS:n kliinisiä piirteitä, mutta lisätutkimuksia tarvitaan.
Varhaisia kliinisiä eroja havaittiin verrattaessa PWS:ää tai AS:ää sairastavia henkilöitä, joilla oli deletoitunut ja ei-deletoitunut status (29), mukaan lukien hypopigmentaatio PWS:ää ja AS:ää sairastavilla henkilöillä, joilla oli deletoitunut 15q11-q13 (30). Myöhemmin raportoitiin korkeampia verbaalisia älykkyysosamääräpisteitä (31) tai psykooseja (32) henkilöillä, joilla oli äidin UPD15, verrattuna deletoituneisiin henkilöihin, joilla oli PWS. Lisäksi Butler ja muut (19) raportoivat alhaisempia adaptiivisia pistemääriä ja enemmän pakko-oireista käyttäytymistä PWS-henkilöillä, joilla oli 15q11-q13-tyypin I deletio verrattuna UPD15:een. Zarcone ym. (33) raportoivat, että henkilöillä, joilla oli PWS ja 15q11-q13-tyypin I deletio, oli enemmän henkilökohtaiseen puhtauteen liittyviä pakko-oireita ja pakkokäyttäytymistä, jota oli vaikea keskeyttää ja joka häiritsi sosiaalisia toimintoja enemmän kuin henkilöillä, joilla oli tyypin II deletio tai UPD15. Angelmanin oireyhtymää koskevassa fenotyypin ja genotyypin välisessä korrelaatiotutkimuksessa Moncla ja muut (34) raportoivat lisääntynyttä kohtausaktiivisuutta niillä, joilla oli suurempi I-luokan deleetio verrattuna niihin, joilla ei ollut deleetioita. Myös mikrokefalia, ataksia, hypotonia ja ruokailuvaikeudet ovat todennäköisempiä deletoituneessa alatyypissä (3). Heillä voi olla vaikeampia kielellisiä häiriöitä erityisesti reseptiivisen kielen ja autististen piirteiden osalta (21, 35). AS:ää sairastavilla henkilöillä, joilla on isän UPD, voi olla parempi reseptiivinen kieli, paremmat motoriset kyvyt ja vähäisempi kohtausten esiintyvyys. Mosaiikkihenkilöillä voi myös olla lievempi fenotyyppi, johon kuuluvat paremmat kielelliset kyvyt, sopeutumiskyky ja vähemmän kohtauksia (36).
Seuraavan sukupolven eksomin tai koko genomin sekvensoinnilla voi myös olla paikkansa PWS:n tai AS:n geneettisessä arvioinnissa, erityisesti niillä henkilöillä, joilla on epätavallisia löydöksiä tai joiden diagnoosi on viivästynyt (esim. UPD15:n segmentaalinen tai totaalinen isodisomia), ja tapauksissa, joissa vanhempien DNA:ta ei ole saatavilla (8). PWS:n ja AS:n geneettisen testauksen käyttöä ja tyyppiä varten kehitettiin kliinikkoa varten uusi geneettisen testauksen vuokaavio, joka on kuvattu ja havainnollistettu graafisessa tiivistelmässä. Tämä vuokaavio voi auttaa geneettisen testauksen tilaamisessa kliinisten oireiden perusteella asianmukaisen diagnoosin, hoidon ja hoidon määrittämiseksi ja mahdollisimman tarkan geneettisen neuvontatiedon antamiseksi muille perheenjäsenille. Ehdotamme, että tätä algoritmia käytetään sekä PWS:n että AS:n diagnostiikan lopulliseen loppuunsaattamiseen. Väitämme, että diagnoosi on epätäydellinen ilman tietoa potilaan erityisestä geneettisestä alatyypistä neuvonnan, ennakoivan ohjauksen, hoidon ja todennäköisten hoitovaihtoehtojen ohjaamiseksi. Molekyyliluokan määrittäminen on tärkeää lääketieteellisen hoidon ja hoidon kannalta ja hyödyllistä kliinikolle, joka osallistuu perheenjäsenten geneettiseen neuvontaan PWS:n tai AS:n osalta.
Tekijöiden panos
MB ja JD osallistuivat käsikirjoituksen laatimiseen, kirjallisuuden tarkasteluun, osallistuivat asiantuntemuksellaan ja muokkasivat käsikirjoitusta.
Kiinnostusristiriita
Tekijät ilmoittavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, jotka voitaisiin tulkita mahdolliseksi eturistiriidaksi.
Kiitokset
Kiitämme Grace Grahamia käsikirjoituksen asiantuntevasta valmistelusta.
1. Bittel DC, Butler MG. Prader-Willin oireyhtymä: kliininen genetiikka, sytogenetiikka ja molekyylibiologia. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Whitman BY. Prader-Willin oireyhtymän hoito. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Angelmanin oireyhtymän kliiniset ja geneettiset näkökohdat. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Fysioterapia. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willin oireyhtymä. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Prader-Willin oireyhtymä: katsaus kliinisiin, geneettisiin ja endokriinisiin löydöksiin. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Prilli-Wady-oireyhtymä. Butler MG. Lihavuuden yhden geenin ja oireyhtymän syyt: havainnollistavia esimerkkejä. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Prader-Willin oireyhtymän imprinting-keskuksen analyysi käyttäen pisaran digitaalista PCR:ää ja seuraavan sukupolven koko eksomin sekvensointia. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Suomenkielinen julkaisu. Hartin S, Hossain WA, Weisensel N, Butler MG. Kolme sisarusta, joilla on Prader-Willin oireyhtymä, joka johtuu imprinting-keskuksen mikrodeleetioista, ja katsaus. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Am J Genetiä. Hassan M, Butler MG. Prader-Willin oireyhtymä ja epätyypilliset submikroskooppiset 15q11-q13-deleetiot, joissa on tai ei ole imprintointivirheitä. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Prader-Willin oireyhtymä ja Schaaf-Yangin oireyhtymä: MAGEL2-geenin kohdalla risteävät neurologiset sairaudet. Taudit. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sairaudet. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi-fenotyyppi, jonka aiheuttaa isän puutos HBII-85 C/D-laatikon pienen nukleaarisen RNA-klusterin osalta. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tekijät, jotka eivät ole vielä saaneet tietää, mitä heistä on jäljellä. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC ym. epätyypillisen 15q11.2-mikrodeletion aiheuttama Prader-Willi-Like-fenotyyppi. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Gennes11020128. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Kromosomi 15:n deleetiot Prader-Willin oireyhtymän syynä. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM19810205303040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Kromosomi 15:n deletion vanhempien alkuperä Prader-Willin oireyhtymässä. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Prader-Willin oireyhtymän molekyyligeneettinen luokittelu: monipaikkainen kohorttitutkimus. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Prader-Willin oireyhtymä: kliiniset ja geneettiset löydökset. Endokrinologi. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Fysioterapia. Butler MG. Prader-Willin oireyhtymä: nykyinen käsitys syistä ja diagnoosista. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Willi-Willi Prilli-oireyhtymä. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Käyttäytymiserot henkilöillä, joilla on Prader-Willin oireyhtymä ja tyypin I tai II deletio ja äidin disomia. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Millaisia tuloksia? Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguise the molecular classes of Angelmanin oireyhtymä. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multi-method assessment of sleep in children with Angelman syndrome: a case-controlled study. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Ks. esim. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Clinical utility gene card for: Anglemanin oireyhtymä. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Suomenkielinen julkaisu. Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Am J Med Genet A (2020) 182:53-63. Buiting K, Williams C, Horsthemke B. Angelmanin oireyhtymä – näkemyksiä harvinaisesta neurogeneettisestä häiriöstä. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Fysiologian laitos. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypical Angelman syndrome due to a mosaic imprinting defect: case reports and review of the literature. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Fysioterapia. Butler MG, Meaney FJ, Palmer CG. Kliininen ja sytogeeninen tutkimus 39 henkilöstä, joilla on Prader-Labhart-Willin oireyhtymä. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hypopigmentaatio: Prader-Labhart-Willi-oireyhtymän yleinen piirre. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Prader-Willin oireyhtymän älylliset piirteet: geneettisten alatyyppien vertailu. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psykoottinen sairaus ihmisillä, joilla on Prader-Willin oireyhtymä kromosomi 15:n äidinpuoleisen uniparentaalisen disomian vuoksi. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstrakti | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. Pakonomaisen käyttäytymisen ja akateemisen suoriutumisen välinen suhde Prader-Willin oireyhtymän kolmessa geneettisessä alatyypissä. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Ks. esim. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Johtopäätökset. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Millainen se on? Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Säilytetty ekspressiivinen kieli mosaiikkisen Angelmanin oireyhtymän fenotyyppisenä määrittäjänä. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar