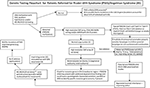
Grafisk resumé. Flowdiagram for genetisk testning af patienter henvist for Prader-Willi syndrom (PWS)/Angelman syndrom (AS). *Udelukke Chr15-translokationer eller inversioner ved rutinemæssige kromosomundersøgelser; overveje andre fedmerelaterede genetiske lidelser; kan kræve fragilt X-syndrom-DNA-screening for FMR1-genrepeatekspansion eller avanceret genetisk testning med næste generations sekventering (NGS) for FMR1 eller andre kandidatgenvarianter ved hjælp af whole-exome-sekventering (WES) eller whole-genome-sekventering (WGS; f.eks. monogene årsager til fedme). **Kan bruges til at kontrollere methyleringsstatus for andre Chr15-imprægnerede gener; ddPCR, droplet digital PCR kan bruges til screening af mosaikisme.
Indledning
Kromosom 15-imprægneringsforstyrrelser omfatter Prader-Willi (PWS) og Angelman (AS) syndromer (1-6) og kromosom 15q-duplikeringer. Diagnosen af PWS eller AS afhænger af den oprindelige forælder og af, om ekspressionen er afvigende begrænset til de moderlige eller faderlige prægetegnede gener. Duplikation 15q skyldes en ekstra kopi af den moderligt afledte 15q11.2-q13-region, som kan føre til kramper, kognitive og adfærdsmæssige problemer, herunder autismespektrumforstyrrelser (ASD), men ikke til en PWS- eller AS-fænotype. PWS opstår som følge af tab af moderligt prægede og faderligt udtrykte gener fra kromosom 15q11-q13-regionen, mens AS skyldes tab af prægede og moderligt udtrykte gener i denne region, hvilket specifikt påvirker UBE3A-genet. På grund af de ansvarlige geners prægethed kan både genetiske og epigenetiske fejl være årsagsskabende.
I 1989 blev det konstateret, at personer med både PWS og ikke-deletionsstatus havde moderens disomi 15 eller begge 15’ere fra moderen, når der blev anvendt polymorfe DNA-markører fra den proximale 15q11-q13-region (7). Senere i midten af 1990’erne blev udviklingen af fluorescerende in situ hybridisering (FISH) DNA-sonder anvendt til at identificere deletioner af 15q11-q13-regionen i både PWS og AS. Methylerings-DNA-testning blev udviklet i denne periode, og der blev set et unormalt methyleringsmønster i PWS og AS. Methylerings-DNA-testning er ~99 % nøjagtig med hensyn til at identificere diagnosen PWS, men identificerer ikke den enkelte PWS-molekylære klasse (2). For AS identificerer DNA-methyleringstest ~80 % af individerne, men skelner heller ikke mellem de molekylære klasser eller påviser en mutation i UBE3A-genet, der forårsager AS.
Microarray-teknologien blev udviklet i begyndelsen til midten af 2000’erne og har fremskyndet det diagnostiske udbytte. Nu omfatter de nye SNP-mikroarrays over to millioner DNA-sonder og er nyttige til påvisning af deletions-subtyper og UPD15-subklasser. Anden teknologi som f.eks. droplet digital PCR (ddPCR) kvantificerer antallet af kopier ved hjælp af DNA-sonder på kromosom 15 og kan diagnosticere genetiske defekter i PWS eller AS (8). Endvidere kan SNP-mikroarrays identificere LOH’er defineret som >8 Mb i størrelse, og når de er til stede på kromosom 15, understøtter de diagnosen maternel disomi 15 eller paternel disomi 15 i tilstedeværelsen af et unormalt DNA-methyleringsmønster for henholdsvis PWS eller AS. Bekræftelse af imprintingdefekt kan kræve ikke kun SNP-mikroarrays for at identificere små mikrodeletioner, men også DNA-prøver fra forældrene med genotypebestemmelse for at identificere tilstedeværelsen af normal (biparental) arv af kromosom 15s, der understøtter tilstedeværelsen af en epimutationsimprintingdefekt i PWS eller AS og dermed påvirker risikoen for recidiv. Differentiering af en IC-mikrodeletion fra en ikke-deletions-epimutationsstatus er klinisk vigtig for familierne, da der er 50 % risiko for gentagelse for yderligere børn, hvis der findes en IC-mikrodeletion hos forældrene (9).
Der er over et dusin gener og transskriptioner i 15q11-q13-regionen, som ser ud til at spille en rolle i forbindelse med forårsagen af PWS og/eller AS. Gener og transkripter, der indgår i området fra det proximale 15q11.2-brudpunkt BP1 og det distale 15q13-brudpunkt BP3, er TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, ikke-koderende RNA’er (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 og HERC2. MRKN3-, MAGEL2-, NDN-, NIPAP1- og SNURF-SNRPN-gener, der er præget af tryk, udtrykkes af faderen og kan, når de er forstyrrede, forårsage træk af PWS . F.eks. kan MAGEL2-genmutationer forårsage neonatal hypotoni, udviklingsforsinkelse, arthrogryposis, autistiske træk, dårlig sugeevne og fedme . Patienter er også blevet rapporteret med træk af PWS som følge af små deletioner af det ikke-kodende SNORD116-transkript (12) og andre lignende deletioner i regionen (10, 13).
Vi fokuserede på AS og PWS i denne rapport, da begge syndromer påvises via DNA-methyleringstest, som gør det muligt at bestemme den aktive forældrenes genallel og stille en endelig diagnose hos personer med PWS og hos de fleste personer med AS (2). DNA-methyleringstest vil imidlertid ikke identificere den molekylære klasse i nogen af syndromerne. Kromosomanalyse med høj opløsning blev udviklet og anvendt i begyndelsen af 1980’erne og blev en standard laboratoriegenetisk baseret test til vurdering af kromosom 15q11-q13-deletionen, der dengang blev identificeret hos størstedelen af patienterne med PWS (14) og senere for AS. Den faderlige oprindelse af 15q11-q13-deletionen blev rapporteret i 1983 (15) og viste sig at være de novo, men størrelsen af 15q11-q13-deletionen eller typen (typisk vs. atypisk) kunne ikke bestemmes. En nøjagtig og tidlig diagnose med identifikation af den molekylære klasse er afgørende, ikke kun for at bekræfte den kliniske diagnose, men også for genetisk rådgivning, for at informere om pleje og behandling og for at styre forventningerne. Med henblik på igangværende kliniske forsøg kan en bedre forståelse af den molekylære ætiologi få indflydelse på mulighederne for patientdeltagelse. Desuden omfatter de forestående forsøg antisense-oligonukleotider til reaktivering af den tavse faderlige kopi af kromosom 15 hos personer med AS.
PWS og AS er komplekse sjældne neuroudviklingsforstyrrelser, der skyldes fejl i genomisk prægning. PWS er anerkendt som den mest almindelige genetiske årsag til livstruende fedme, hvis den forbliver ukontrolleret (2, 4, 6). Der er tre anerkendte PWS-molekylære klasser, herunder en faderlig 15q11-q13-deletion på ca. 5-6 Mb (60 % af tilfældene) og moderlig disomi 15 (UPD15), hvor begge kromosomer 15 nedarves fra moderen (36 %), der stammer fra trisomi 15 med tab af det faderlige kromosom 15 i den tidlige graviditet, hvilket fører til to kromosomer 15 fra moderen (16). Den tredje klasse er en defekt i imprintingcentret. Hvis en mikrodeletion eller epimutation af imprintingcentret (IC), som kontrollerer udtryksstatus for udvalgte imprintede gener på kromosom 15, er til stede på den faderlige allel, opstår der PWS. Denne imprintingdefekt ses hos 4 % af alle personer med PWS (8, 16). De fleste tilfælde af PWS er sporadiske med en tilnærmelsesvis ligelig fordeling mellem etniske grupper og køn. Den anslåede prævalens af PWS er en ud af 10.000 til en ud af 30.000 (2). Antallet af personer på verdensplan med PWS menes at være ~400.000 med ca. 20.000 personer i USA (2, 17).
PWS er karakteriseret ved infantil hypotoni, dårlig sutterefleks med ernæringsvanskeligheder, kort statur med små hænder og fødder, hypogonadisme sekundært til hormonmangel, let intellektuel funktionsnedsættelse, adfærdsproblemer og hyperfagi ofte med debut mellem 6 og 8 års alderen, der fortsætter ind i voksenalderen og resulterer i fedme, hvis der ikke er miljøkontrol. I spædbarnsalderen ses karakteristiske kraniofaciale træk, herunder en smal bifrontal diameter, strabismus, lille opadvendt næse med en tynd overlæbe og nedadvendte mundvigene, klæbrigt spyt og emaljehypoplasi (2, 4, 6, 18). Kognitionen er generelt nedsat på baggrund af familiebaggrunden, og adfærdsproblemer, der begynder i barndommen, omfatter selvskade (hudplukning), udbrud, stædighed og raserianfald med psykiatriske problemer, der opstår i denne periode eller senere i ungdomsårene eller i den unge voksenalder (2). Adfærdsproblemer omfatter angst, humørsvingninger, psykoser og autisme, der kan korrelere med specifikke genetiske undertyper eller molekylære klasser af PWS (19).
Historisk set er PWS opdelt i to kliniske stadier med manglende trivsel i spædbarnsalderen, der repræsenterer det første kliniske stadium, og hyperfagi med begyndende fedme, der repræsenterer det andet stadium (2). Senere er der blevet beskrevet ernæringsfaser for denne fedmerelaterede genetiske lidelse, og de omfatter: Fase 0 med nedsat fosterbevægelse og væksthæmning in utero, efterfulgt af fase 1 i forbindelse med hypotoni, manglende trivsel med ernæringsvanskeligheder, fase 2, der begynder ved ~2 års alderen, hvor vægtøgning først konstateres, og fase 3, hvor manglende mæthed ledsages af fødevaresøgning og hyperfagi, der fører til fedme, hvis den ikke kontrolleres eksternt. Fase 3 begynder omkring 6-8 års alderen (20).
Angelman syndrom er karakteriseret ved udviklingsforsinkelse, der ofte ikke er synlig før omkring 6 måneders alderen og efterfølgende indtræden af ofte svært kontrollerbare anfald, tremor, bredbaset gang og ataksi med en karakteristisk glad opførsel (3). Der er fire anerkendte molekylære mekanismer for AS: de novo maternelle deletioner af kromosom 15q11-q13 (70-80 %); mutationer i det moderligt nedarvede UBE3A-gen (10-20 %); faderlig disomi 15 (3-5 %); og imprintingdefekter (3-5 %) inden for 15q11-q13-regionen, der ændrer udtrykket af det forårsagende UBE3A-gen (21).
Individer med AS bliver ofte ikke bemærket af læger før ~6 måneders alderen, når forsinkelser i udviklingen især forsinket motorisk udvikling rapporteres. På dette tidspunkt kan forældrene genkende den glade opførsel, der omfatter hyppig latter, smil og ophidselse. Der rapporteres om et nedsat søvnbehov hos >80 % af personer med AS (22). De udvikler ofte anfald i alderen 1-3 år (23). Epilepsi kan være uhelbredelig og har et karakteristisk udseende på EEG, der beskrives som en øget deltaeffekt med en karakteristisk trifasisk bølge. Personer med AS beskrives som ataktiske i deres bevægelser og gang (24, 25). Mikrocefali kan udvikle sig i en alder af ~2 år. Stereotypisk adfærd omfatter en kærlighed til vand og knitrende papir, og personer med AS er karakteristisk non-verbale og kategoriseret som alvorligt intellektuelt handicappede. Det er imidlertid bemærkelsesværdigt, at personer med AS har færdigheder, der ikke er godt fanget på de i øjeblikket tilgængelige objektive neuropsykologiske tests. De har stærke evner til at manipulere elektronik, men adfærd kan være udfordrende og omfatte angst med kort opmærksomhedsspændvidde.
Da patienter med PWS eller AS kan have forskellige fænotyper afhængigt af molekylærklasse, og da der findes potentielle behandlings- og overvågningsmetoder for hver af dem, er der behov for et logisk flowdiagram til bestilling af genetiske test af den kliniker, der evaluerer disse patienter. Fokus i vores rapport er at beskrive de kliniske og genetiske fund af disse to genomiske prægningsforstyrrelser og at illustrere de genetiske testmuligheder, der er tilgængelige i kliniske omgivelser, og den rækkefølge, i hvilken de forskellige genetiske tests kan opnås mest produktivt.
Laboratoriegenetisk erfaring med kromosom 15 Imprinting Disorders
Prader-Willi syndrom
For at tjene som et eksempel på betydningen af SNP-mikroarray-test med høj opløsning blev en stor multisite-kohorte på 510 deltagere med genetisk bekræftet PWS rekrutteret i USA og grupperet i tre molekylære klasser. De blev yderligere karakteriseret som 15q11-q13-deletionssubtyper, maternel disomi 15-subklasser og imprintingcenterdefekter (16). I denne største rapporterede PWS-kohorte blev 303 individer fundet at have 15q11-q13-deletionen (60 % af tilfældene) sammensat af 118 individer (38,9 %) med den større typiske 15q11-q13-type I-deletion, der involverer kromosom 15q11-q13-brudpunkterne BP1 og BP3, og 165 individer (54.5 %) havde den mindre typiske typiske 15q11-q13 type II-deletion, der involverer brudpunkterne BP2 og BP3, med 20 personer med en atypisk deletion, der er større eller mindre end den typiske 15q11-q13-deletion (6,6 %). Hos personer, der er identificeret som havende en deletion af kromosom 15, er det vigtigt at overveje, om der kan være en balanceret translokation til stede hos probandens far, da dette øger risikoen for gentagelse af PWS hos faderens afkom. For maternel disomi 15 havde 185 personer (36 %) maternel uniparental disomi 15 (UPD15) med 13 personer (12,5 %) med total isodisomi af hele kromosom 15 på grund af fejl i moderens meiose II; 60 (57,7 %) viste segmentel isodisomi fra crossover-hændelser i moderens meiose I og 31 viste heterodisomi (29,8 %), mens 81 personer ikke havde SNP-mikroarrayanalyse og moderens disomi 15-klassificering bestemt. Med hensyn til PWS-indtryksdefekter blev der fundet 22 individer (4 %), hvoraf 13 (76,5 %) havde en ikke-deletionsepimutationsstatus, fire individer (23,5 %) havde en mikrodeletion af indtrykscentret, mens de resterende fem individer ikke havde fået fastlagt en type indtryksdefekt. I en beslægtet undersøgelse blev der foretaget en yderligere analyse af imprintingdefekter i PWS af Hartin et al. (8) ved hjælp af droplet digital PCR og next-generation whole-exome-sekventering i en separat PWS-kohorte af 15 ubeslægtede patienter, og det blev konstateret, at to personer eller 13 % havde en mikrodeletionsdefekt i imprintingcentret. Hos de 60 individer med segmental isodisomi 15 rapporteret af Butler et al. (16) var den samlede gennemsnitlige størrelse af tab af heterozygositet (LOH) 25,1 Mb med et interval på 5-67,4 Mb og en gennemsnitlig størrelse på 16,4 Mb for individuelle LOH’er. 32 individer havde ét LOH-segment, 25 individer havde to segmenter, og tre individer havde tre segmenter. De mest almindelige LOH-steder var den proximale 15q11-q13-region og den distale 15q26-region, herunder 15q12- og 15q26.1-båndene som de hyppigst registrerede.
Anstedeværelsen af moderens UPD15- og specifik underklassebestemmelse (segmental eller total isodisomi) kan påvirke diagnosen og overvågning af medicinsk behandling, da der kan være en anden genetisk tilstand til stede, hvis moderen er bærer af en recessiv genallel placeret i LOH-regionen, hvilket fører til to identiske kopier. Der findes hundredvis af potentielt sygdomsfremkaldende gener på kromosom 15, og disse sygdomme bør kontrolleres eller overvåges nøje hos personer med segmental eller total isodisomi af kromosom 15. Et forslag til et flowdiagram for genetisk testning til identifikation af de forskellige molekylære klasser for både PWS- og AS-patienter kan ses i Graphical Abstract.
Angelman syndrom
Fire anerkendte molekylære klasser er blevet identificeret i AS, som kan kategoriseres efter indvirkningen på methyleringen af kromosom 15-regionen. Den mest almindelige undertype er en deletion af det maternelle 15q11.2-q13-regionen, som ligeledes ses af faderlig oprindelse i PWS og findes hos ~70% af personer med AS (21). I AS er den typiske klasse II-deletion dog mere almindelig. Denne typiske mindre klasse II-deletion er oftest ca. 5 Mb stor fra BP2-BP3 og er til stede i 50 % af AS-deletionstilfælde. Klasse I-deletioner er 5-7 Mb store og omfatter BP1-BP3 (40 % af deletionstilfælde). Atypiske deletioner kan strække sig fra BP1 eller BP2-BP4 eller til mere fjerntliggende brudpunkter. Hos personer med en deletion på den maternelle kopi af kromosom 15 skal man overveje, om der er tegn på kromosomale mikroarray, der viser forstyrrelser, som tyder på, at der kan være tale om en translokation hos moderen. Dette øger risikoen for gentagelse af AS hos fremtidige moderlige afkom. Uniparental paternal disomi 15 udgør 5-7 % af personer med AS. Imprintingdefekter tegner sig for 3-5 % af personer med AS og skyldes defekter i imprinting-kontrolcentret, som Buiting et al. har opsummeret (26). Hos personer med en defekt i imprinting-kontrolcentret undlader den epigenetiske markering i kimlinjen at skifte korrekt fra et faderligt mønster med tavs UBE3A-ekspression til at tillade et moderligt ekspressionsmønster på UBE3A-genet. I helt op til 50 % af de rapporterede tilfælde kan der identificeres en mutation i imprinting-kontrolcentret. Mosaiktilfælde af defekter i imprintingcentret, hvor en procentdel af cellerne mangler udtryk for 15q11.2-q13-regionen, er rapporteret og kan være mere almindelige end tidligere antaget (27). Den sidste genetiske defekt i AS påvirker ikke resultaterne af DNA-methyleringstest, men skyldes en mutation i det moderligt nedarvede UBE3A-gen. Mutationer i dette gen tegner sig for 11 % af AS-tilfældene (28). En UBE3A-mutation kan være moderligt arvelig, og derfor er det indiceret at foretage målrettet testning af patientens mor for at udelukke en 50 % risiko for recidiv hos hendes fremtidige afkom. Hvis mutationen vurderes at være arvelig, anbefaler vi, at man overvejer at teste patientens morfar fra moderen, da dette kan have konsekvenser for moderens tantes fremtidige børn.
Diskussion
Medicinsk behandling af PWS og AS bør ledes af et tværfagligt team i spædbarnsalderen. Både spædbørn med PWS (mere almindeligt) og AS kan have manglende trivsel. En diætist spiller en vigtig rolle i plejen i første omgang for at afhjælpe manglende trivsel og senere i barndommen for at undgå fedme med kostintervention med restriktion og brug af motionsprogrammer (hvilket er en bekymring, der er bemærket mere almindeligt for PWS, men nu anerkendt i AS hos nogle personer). Der er behov for kliniske genetikere, ortopædkirurgiske specialister, læger i primær sundhedspleje, specialiserede terapeuter inden for beskæftigelse (OT), fysioterapi (PT) og tale (SLP), eksperter inden for mental sundhed, søvnspecialister, eksperter inden for mental sundhed og endokrinologer for at løse de mange sundhedsproblemer, der kan opstå i PWS. Et AS-team omfatter kliniske genetikere, neurologer, specialiserede terapeuter til PT-, OT- og SLP-tjenester, søvnspecialister, gastroenterologi, fysisk medicin og rehabilitering, ortopædkirurgi og eksperter inden for mental sundhed. For PWS er passende medicinsk behandling, styring og rådgivning mål for at kontrollere vægtøgning og overvåge og behandle tilknyttede comorbiditetstilstande, adfærd og psykiatriske problemer. Vækstmangel og andre hormonmangler, der er almindelige ved denne lidelse, kræver behandling. Streng kontrol af kosten med fødevaresikkerhed og et styret rutinemiljø med regelmæssig motion er vigtige strategier til at kontrollere hyperfagi, fedme og relaterede komplikationer, som er nødvendige hele livet igennem. AS kræver tidlig indgriben, herunder kendskab til specialiserede terapeutiske interventioner som f.eks. forstærkende og hjælpemidler til kommunikation og et styrkelsesprogram med intensive udviklingsøvelser og aktiviteter for at nå det maksimale potentiale (f.eks. SPIDER), tidlig behandling med benzodiazepiner mod anfald og diætbehandling som f.eks. brug af ketogen diæt. Maksimering af alle aspekter af plejen, herunder søvnforstyrrelser og forstoppelse, har stor indflydelse på anfaldskontrol. Et specialiseret center, der er bekendt med de indviklede og unikke aspekter af disse lidelser, kan påvirke resultatet.
Tidlig diagnose er afgørende for at sikre tidlig intervention for både PWS og AS. For PWS bør der stilles en tidlig diagnose i spædbarnsalderen for at iværksætte væksthormonbehandling, håndtere ernæringsproblemer, fedme, hormonmangel, udviklingsforsinkelser og adfærdsmæssige problemer. Ved diagnosticering af AS sikres også tidlig behandling, som har indflydelse på udviklingsresultaterne, samt anfaldsprofylakse, herunder forberedelse med passende benzodiazepiner. Andre interventioner, der kan vise sig at være gavnlige, omfatter specialiserede diæter til personer med AS som f.eks. ketogen diæt eller behandling med lavt glykæmisk indeks (LGIT). Tidlig diagnose kan også sænke udgifterne til medicinsk behandling ved at forebygge længerevarende hospitalsindlæggelser i forbindelse med ernæringsproblemer hos personer med PWS og kramper hos børn med AS.
Identificering af den molekylære klasse for PWS eller AS med avancerede genetiske test såsom SNP-mikroarrays med høj opløsning vil muliggøre en mere præcis diagnose, hvilket vil føre til bedre indikatorer for prognosen og mere præcis genetisk rådgivning af familiemedlemmer. SNP-mikroarrays med høj opløsning, FISH-analyse, methyleringsspecifik multiplex-ligeringssondeamplifikation (MS-MLPA) og/eller kromosom 15-genotypebestemmelse er alle nyttige til bestemmelse af 15q11-q13-deletioner. SNP-mikroarrays med høj opløsning kan identificere deletions-subtyperne (typiske og atypiske) i både PWS og AS og UPD15-subklasser (segmental isodisomi og total isodisomi). Heterodisomi-subtypen af UPD- og IC-defekter (mikrodeletion og epimutation) i både PWS og AS kan kræve yderligere diagnostisk udredning, som illustreret i det grafiske resumé. Subtypen eller klasserne påvirker diagnosen, den potentielle risiko for gentagelse for familiemedlemmer, prognosen og overvågningen af andre genetiske tilstande og højrisikoegenskaber i forbindelse med den molekylære klasse. F.eks. er autistiske træk og psykoser mere almindelige hos personer med PWS og moderdiskomi 15 og kan være forbundet med de specifikke UPD15-subklasser. De med de større klasse I-deletioner i AS er mere tilbøjelige til at udvikle svært behandlelige anfald og mikrocefali.
Et flowdiagram for genetisk testning, der omfatter de testmuligheder, der er til rådighed, herunder dem, der historisk er blevet anvendt til både PWS og AS, er anført i det grafiske resumé. Testning for PWS eller AS begynder ofte med DNA-methylering, og hvis den er unormal, går man videre til andre genetiske testmetoder, herunder højopløselige SNP-mikroarrays eller MS-MLPA-analyser, baseret på tilgængelighed for klinikere og familier i deres kliniske omgivelser. Der vil fortrinsvis blive bestilt et SNP-array med høj opløsning, som er let og kommercielt tilgængeligt i den vestlige sundhedssektor. Næste generations sekventering (NGS) af exomet (eller hele genomet) er også tilgængelig for klinikere, men droplet digital PCR (ddPCR) er i øjeblikket forskningsbaseret (14). SNP arrays kan identificere specifikke molekylære klasser i størstedelen af patienter, der præsenterer sig med træk af PWS (ca. 85 % af tilfældene) eller AS (ca. 80 %), mens de resterende patienter vil have brug for yderligere testning som beskrevet i grafisk resumé. Specifikke avancerede genetiske test (f.eks. ddPCR) kan være tilstrækkeligt følsomme til at kvantificere mosaikisme og kan identificere en diagnose i en stor delmængde af individer med mildere kliniske træk ved PWS og AS, men der er behov for mere forskning.
Der blev fundet tidlige kliniske forskelle ved sammenligning af dem med PWS eller AS med deletionsstatus vs. ikke-deletionsstatus (29), herunder hypopigmentering hos dem med PWS og AS med 15q11-q13-deletion (30). Senere blev der rapporteret højere verbale IQ-scoringer (31) eller psykose (32) hos dem med moderens UPD15 i forhold til deletion hos personer med PWS. Endvidere rapporterede Butler et al. (19) lavere adaptive scorer og mere obsessiv-kompulsiv adfærd hos personer med PWS med 15q11-q13 type I-deletion sammenlignet med UPD15. Zarcone et al. (33) rapporterede, at personer med PWS og 15q11-q13 type I-deletion havde flere tvangstanker om personlig renlighed og tvangsadfærd, som var vanskelig at afbryde og i højere grad forstyrrede sociale aktiviteter end personer med type II-deletion eller UPD15. I en fænotype-genotype korrelationsundersøgelse af Angelman syndrom rapporterede Moncla et al. (34) om øget anfaldsaktivitet hos dem med den større klasse I-deletion sammenlignet med dem uden deletion. Mikrocefali, ataksi, hypotoni og ernæringsvanskeligheder er også mere sandsynlige i deletionsundertypen (3). De kan have mere alvorlige sproglige forstyrrelser, især receptivt sprog og autistiske træk (21, 35). Personer med AS med faderlig UPD kan have forbedret receptivt sprog, forbedrede motoriske evner og en nedsat prævalens af anfald. Mosaikindivider kan også have en mildere fænotype, herunder forbedrede sproglige evner, adaptiv funktion og færre anfald (36).
Næste generations exom- eller helgenomsekventering kan også have en plads i genetiske evalueringer i PWS eller AS, især hos de individer, der præsenterer sig med usædvanlige fund eller forsinket diagnose (f.eks. UPD15 segmental eller total isodisomi) og i tilfælde, hvor forældrenes DNA ikke er tilgængeligt (8). For at tage fat på brugen og typen af genetisk testning for PWS og AS blev der udviklet et nyt flowdiagram for genetisk testning til klinikeren, som beskrevet og illustreret i Graphical Abstract. Dette flowdiagram kan hjælpe med at bestille genetisk testning baseret på den kliniske præsentation for at bestemme passende diagnose, håndtering og behandling og for at give de mest præcise genetiske rådgivningsoplysninger til andre familiemedlemmer. Vi foreslår, at denne algoritme anvendes til endeligt at færdiggøre den diagnostiske undersøgelse af både PWS og AS. Vi hævder, at diagnosen er ufuldstændig uden kendskab til patientens specifikke genetiske subtype for at kunne vejlede om rådgivning, foregribende vejledning, håndtering og sandsynlige behandlingsmuligheder. Den molekylære klassebestemmelse er vigtig for medicinsk pleje og behandling og nyttig for den kliniker, der er involveret i genetisk rådgivning af familiemedlemmer for PWS eller AS.
Author Contributions
MB og JD bidrog til udarbejdelse af manuskriptet, gennemgang af litteraturen, bidrog med deres ekspertise og redigerede manuskriptet.
Funding
Vi anerkender National Institute of Child Health and Human Development grant number HD02528.
Interessekonflikter
Forfatterne erklærer, at forskningen blev udført uden kommercielle eller finansielle relationer, der kunne opfattes som en potentiel interessekonflikt.
Akkreditering
Vi anerkender Grace Graham for ekspertudarbejdelse af manuskriptet.
1. Bittel DC, Butler MG. Prader-Willi syndrom: klinisk genetik, cytogenetik og molekylærbiologi. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG, Lee PDK, Lee PDK, Whitman BY. Håndtering af Prader-Willi-syndromet. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Kliniske og genetiske aspekter af Angelman syndrom. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willi syndrom. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrom: en gennemgang af kliniske, genetiske og endokrine fund. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Enkeltgen og syndromiske årsager til fedme: illustrative eksempler. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Analyse af Prader-Willi-syndromets imprintingcenter ved hjælp af droplet digital PCR og næste generations whole-exome-sekventering. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Tre søskende med Prader-Willi syndrom forårsaget af imprinting center mikrodeletioner og gennemgang. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Prader-Willi syndrom og atypiske submikroskopiske submikroskopiske 15q11-q13 deletioner med eller uden imprintingdefekter. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Prader-Willi syndrom og Schaaf-Yang syndrom: neurodevelopmental diseases intersecting at the MAGEL2 gene. Sygdomme. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi-fænotype forårsaget af faderlig mangel for HBII-85 C/D-boks lille nukleolær RNA-klynge. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-lignende fænotype forårsaget af en atypisk 15q11.2-mikrodeletion. Genes (Basel). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deletioner af kromosom 15 som en årsag til Prader-Willi-syndromet. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Forældrenes oprindelse af kromosom 15-deletion ved Prader-Willi syndrom. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Molekylær genetisk klassifikation af Prader-Willi syndrom: en kohorteundersøgelse på flere forskellige steder. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Prader-Willi syndrom: kliniske og genetiske fund. Endokrinolog. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Butler MG. Prader-Willi syndrom: nuværende forståelse af årsag og diagnose. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Adfærdsforskelle blandt personer med Prader-Willi syndrom og type I eller type II deletion og maternel disomi. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Ernæringsfaser i Prader-Willi syndrom. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrom. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Multimetodevurdering af søvn hos børn med Angelman-syndrom: en case-kontrolleret undersøgelse. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Klinisk nytte genkort for: Angleman syndrom. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Adfærd og neuropsykiatriske manifestationer i Angelman syndrom. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. En oversigt over sundhedsspørgsmål og udvikling i en stor klinisk kohorte af børn med Angelman syndrom. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelman syndrom – indsigt i en sjælden neurogenetisk lidelse. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Atypisk Angelman-syndrom som følge af en mosaisk imprintingfejl: case reports and review of the literature. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neuroterapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Klinisk og cytogen undersøgelse af 39 personer med Prader-Labhart-Willi syndrom. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.132023030307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Hypopigmentering: et almindeligt træk ved Prader-Labhart-Willi syndrom. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Intellektuelle karakteristika ved Prader-Willi syndrom: sammenligning af genetiske undertyper. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psykotisk sygdom hos personer med Prader-Willi syndrom på grund af kromosom 15 maternel uniparental disomi. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. Forholdet mellem tvangsadfærd og akademisk præstation på tværs af de tre genetiske undertyper af Prader-Willi syndrom. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Fænotype-genotype korrelation i 20 deletions- og 20 ikke-deletions Angelman syndrom patienter. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identifikation af nye deletioner af 15q11q13 i Angelman-syndromet ved array-CGH: molekylær karakterisering og genotype-fænotypekorrelationer. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Bevaret ekspressivt sprog som en fænotypisk determinant for mosaisk Angelman syndrom. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar