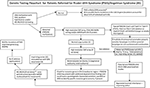
Resumen gráfico. Diagrama de flujo de las pruebas genéticas para los pacientes remitidos por el síndrome de Prader-Willi (SPW)/síndrome de Angelman (SA). *Descartar translocaciones o inversiones de Chr15 mediante estudios cromosómicos rutinarios; considerar otros trastornos genéticos relacionados con la obesidad; puede requerir el cribado del ADN del síndrome del cromosoma X frágil para la expansión de la repetición del gen FMR1 o pruebas genéticas avanzadas con secuenciación de próxima generación (NGS) para FMR1 u otras variantes genéticas candidatas utilizando la secuenciación del exoma completo (WES) o la secuenciación del genoma completo (WGS; por ejemplo, causas monogénicas de la obesidad). **Puede utilizarse para comprobar el estado de metilación de otros genes impresos del cromosoma 15; la ddPCR, la PCR digital en gotas puede utilizarse para el cribado de mosaicismo.
Introducción
Los trastornos de impresión del cromosoma 15 incluyen los síndromes de Prader-Willi (SPW) y Angelman (AS) (1-6) y las duplicaciones del cromosoma 15q. El diagnóstico del SPW o del SA depende del padre de origen y de si la expresión se limita de forma aberrante a los genes impresos maternos o paternos. La duplicación 15q está causada por una copia adicional de la región 15q11.2-q13 de origen materno que puede dar lugar a convulsiones, problemas cognitivos y de comportamiento, incluido el trastorno del espectro autista (TEA), pero no a un fenotipo de SPW o SA. El SPW surge de la pérdida de genes impresos por la madre y expresados por el padre en la región cromosómica 15q11-q13, mientras que el SA está causado por la pérdida de genes impresos y expresados por la madre en esta región, afectando específicamente al gen UBE3A. Debido a la naturaleza impresa de los genes responsables, tanto los errores genéticos como los epigenéticos pueden ser causantes.
En 1989, se descubrió que las personas con SPW y sin deleción tenían disomía materna 15 o ambas 15 de la madre cuando se utilizaron marcadores de ADN polimórficos de la región proximal 15q11-q13 (7). Más tarde, a mediados de los 90, el desarrollo de sondas de ADN de hibridación in situ fluorescente (FISH) se utilizaron para identificar deleciones de la región 15q11-q13 tanto en el SPW como en el SA. Las pruebas de metilación del ADN se desarrollaron durante este periodo y se observó un patrón de metilación anormal en el SPW y en el SA. El test de metilación del ADN tiene una precisión del ~99% en la identificación del diagnóstico del SPW, pero no identifica la clase molecular individual del SPW (2). Para el SA, la prueba de metilación del ADN identifica el ~80% de los individuos, pero de nuevo no distingue entre las clases moleculares o detecta una mutación en el gen UBE3A que causa el SA.
La tecnología de microarrays se desarrolló a principios y mediados de la década de 2000 y avanzó el rendimiento del diagnóstico. Ahora los nuevos microarrays de SNP incluyen más de dos millones de sondas de ADN y son útiles para detectar subtipos de deleción y subclases de UPD15. Otra tecnología, como la PCR digital en gotas (ddPCR), cuantifica el número de copias utilizando sondas de ADN del cromosoma 15 y puede diagnosticar defectos genéticos en el SPW o el SA (8). Además, los microarrays de SNP pueden identificar LOHs definidos como >8 Mb de tamaño y cuando están presentes en el cromosoma 15 apoyan el diagnóstico de disomía materna 15 o paterna 15 en presencia de un patrón de metilación de ADN anormal para el SPW o el SA, respectivamente. La confirmación del defecto de impronta puede requerir no sólo microarrays de SNP para identificar pequeñas microdeleciones, sino también muestras de ADN paterno con genotipado para identificar la presencia de una herencia normal (biparental) del cromosoma 15 que apoye la presencia de un defecto de impronta por epimutación en el SPW o en el SA, impactando así en los riesgos de recurrencia. La diferenciación de una microdeleción IC de un estado de epimutación no-deleción es clínicamente importante para las familias ya que existe un riesgo de recurrencia del 50% para los hijos adicionales si se encuentra una microdeleción IC en el padre (9).
Hay más de una docena de genes y transcripciones en la región 15q11-q13 que parecen desempeñar un papel en la causalidad del SPW y/o el SA. Los genes y transcritos incluidos en el área desde los puntos de rotura proximal 15q11.2 BP1 y el punto de rotura distal 15q13 BP3 son TUBGCP5, CYFIP1, NIPA1, NIPA2, MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, ARN no codificante (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2, y HERC2. Los genes impresos MRKN3, MAGEL2, NDN, NIPAP1 y SNURF-SNRPN se expresan paternalmente y cuando se alteran pueden causar características del SPW. Por ejemplo, las mutaciones del gen MAGEL2 pueden causar hipotonía neonatal, retraso en el desarrollo, artrogriposis, rasgos autistas, mala succión y obesidad. También se han descrito pacientes con características de SPW como resultado de pequeñas deleciones del transcrito no codificante SNORD116 (12) y otras deleciones similares en la región (10, 13).
Nos centramos en el SA y el SPW en este informe ya que ambos síndromes se detectan a través de las pruebas de metilación del ADN, que permiten determinar el alelo activo del gen parental y el diagnóstico definitivo en individuos con SPW y en la mayoría de los individuos con SA (2). Sin embargo, las pruebas de metilación del ADN no identifican la clase molecular en ninguno de los dos síndromes. El análisis cromosómico de alta resolución se desarrolló y utilizó a principios de la década de 1980 y se convirtió en una prueba estándar de laboratorio basada en la genética para evaluar la deleción del cromosoma 15q11-q13 identificada en la mayoría de los pacientes con SPW en ese momento (14) y posteriormente para el SA. El origen paterno de la deleción 15q11-q13 fue reportado en 1983 (15) y se encontró que era de novo pero el tamaño de la deleción 15q11-q13 o el tipo (típico vs. atípico) no pudo ser determinado. El diagnóstico preciso y temprano con la identificación de la clase molecular es esencial no sólo para confirmar el diagnóstico clínico sino también para el asesoramiento genético, para informar sobre la atención y el tratamiento y para guiar las expectativas. Con la intención de realizar ensayos clínicos en curso, una mejor comprensión de la etiología molecular puede repercutir en las oportunidades de participación de los pacientes. Además, los ensayos inminentes incluyen oligonucleótidos antisentido para reactivar la copia paterna silenciada del cromosoma 15 en individuos con SA.
El SPW y el SA son trastornos complejos y raros del neurodesarrollo debidos a errores en la impronta genómica. El SPW está reconocido como la causa genética más común de obesidad potencialmente mortal, si no se controla (2, 4, 6). Hay tres clases moleculares de SPW reconocidas, incluyendo una deleción paterna 15q11-q13 de unos 5-6 Mb de tamaño (60% de los casos) y la disomía materna 15 (UPD15) en la que ambos cromosomas 15 se heredan de la madre (36%) originada por la trisomía 15 con la pérdida del cromosoma 15 paterno al principio del embarazo que lleva a dos cromosomas 15 de la madre (16). La tercera clase es un defecto del centro de impronta. Si una microdeleción o epimutación del centro de impronta (IC), que controla el estado de expresión de determinados genes impresos en el cromosoma 15, está presente en el alelo paterno, se produce el SPW. Este defecto de impronta se observa en el 4% de los individuos con SPW (8, 16). La mayoría de los casos de SPW son esporádicos con una equidad aproximada entre grupos étnicos y sexo. La prevalencia estimada del SPW es de uno en 10.000 a uno en 30.000 (2). Se cree que el número de individuos con SPW en todo el mundo es de unos 400.000, de los cuales unos 20.000 viven en EE.UU. (2, 17).
El SPW se caracteriza por hipotonía infantil, un reflejo de succión deficiente con dificultades de alimentación, baja estatura con manos y pies pequeños, hipogonadismo secundario a deficiencias hormonales, discapacidad intelectual leve, problemas de comportamiento e hiperfagia, a menudo con un inicio entre los 6 y 8 años de edad que persiste en la edad adulta y da lugar a la obesidad si no hay controles ambientales. Durante la infancia, se observan rasgos craneofaciales característicos que incluyen un diámetro bifrontal estrecho, estrabismo, nariz pequeña y respingona con un labio superior delgado y comisuras de la boca dobladas hacia abajo, saliva pegajosa e hipoplasia del esmalte (2, 4, 6, 18). La cognición suele estar reducida en función de los antecedentes familiares y los problemas de comportamiento que comienzan en la infancia incluyen autolesiones (arrancarse la piel), arrebatos, terquedad y rabietas, con problemas psiquiátricos que aparecen durante esta época o más tarde en la adolescencia o en la juventud (2). Los problemas de comportamiento incluyen ansiedad, trastornos del estado de ánimo, psicosis y autismo que pueden correlacionarse con subtipos genéticos o clases moleculares específicas del SPW (19).
Históricamente, el SPW se divide en dos etapas clínicas, representando el fracaso del crecimiento durante la infancia la primera etapa clínica y la hiperfagia con el inicio de la obesidad la segunda etapa (2). Posteriormente, se han descrito fases nutricionales para este trastorno genético relacionado con la obesidad e incluyen: La fase 0 con disminución de los movimientos fetales y retraso del crecimiento en el útero, seguida de la fase 1 relacionada con la hipotonía, el retraso en el desarrollo con dificultad para alimentarse, la fase 2 que comienza a los ~2 años de edad cuando se observa por primera vez el aumento de peso y la fase 3 cuando la falta de saciedad se acompaña de búsqueda de comida e hiperfagia que conduce a la obesidad, si no se controla externamente. La fase 3 comienza en torno a los 6-8 años de edad (20).
El síndrome de Angelman se caracteriza por un retraso en el desarrollo que a menudo no se manifiesta hasta los 6 meses de edad y la posterior aparición de convulsiones, a menudo difíciles de controlar, temblores, marcha ancha y ataxia con un comportamiento alegre característico (3). Existen cuatro mecanismos moleculares reconocidos del SA: deleciones maternas de novo del cromosoma 15q11-q13 (70-80%); mutaciones del gen UBE3A heredado por la madre (10-20%); disomía 15 paterna (3-5%); y defectos de impronta (3-5%) dentro de la región 15q11-q13 que alteran la expresión del gen UBE3A causante (21).
Los individuos con SA a menudo no son percibidos por los profesionales médicos hasta los 6 meses de edad, cuando se registran retrasos en el desarrollo, especialmente en el desarrollo motor. Para entonces, los padres pueden reconocer el comportamiento alegre que incluye risas frecuentes, sonrisas y excitabilidad. Se informa de una disminución de la necesidad de dormir en >80% de los individuos con SA (22). A menudo desarrollan convulsiones entre los 1 y 3 años de edad (23). La epilepsia puede ser intratable y tiene un aspecto característico en el EEG descrito como un aumento de la potencia delta con una onda trifásica característica. Los individuos con EA se describen como atáxicos en sus movimientos y en la marcha (24, 25). La microcefalia puede desarrollarse a los 2 años de edad. Los comportamientos estereotipados incluyen el amor por el agua y el papel arrugado, y los individuos con SA son característicamente no verbales y se clasifican como discapacitados intelectuales graves. Sin embargo, cabe destacar que los individuos con SA tienen habilidades que no están bien recogidas en las pruebas neuropsicológicas objetivas disponibles actualmente. Tienen fuertes habilidades en la manipulación de la electrónica, pero los comportamientos pueden ser desafiantes e incluyen la ansiedad con períodos de atención cortos.
Como los pacientes con SPW o AS pueden presentar fenotipos variables dependiendo de la clase molecular y porque existen enfoques potenciales de tratamiento y vigilancia para cada uno, se necesita un diagrama de flujo lógico para ordenar las pruebas genéticas por el clínico que evalúa a estos pacientes. El objetivo de nuestro informe es describir los hallazgos clínicos y genéticos de estos dos trastornos de impronta genómica e ilustrar las opciones de pruebas genéticas disponibles en el entorno clínico y el orden en el que se pueden obtener las diferentes pruebas genéticas de forma más productiva.
Experiencia en genética de laboratorio en los trastornos de impronta del cromosoma 15
Síndrome de Prader-Willi
Para servir de ejemplo de la importancia de las pruebas de microarrays de SNP de alta resolución, se reclutó en Estados Unidos una gran cohorte multisitio de 510 participantes con SPW confirmado genéticamente y se agruparon en tres clases moleculares. Se caracterizaron además como subtipos de deleción 15q11-q13, subclases de disomía materna 15 y defectos del centro de imprinting (16). En esta cohorte más grande de SPW, se encontró que 303 individuos tenían la deleción 15q11-q13 (60% de los casos) compuesto por 118 individuos (38.9%) que tenían la deleción típica 15q11-q13 Tipo I más grande que involucraba los puntos de ruptura BP1 y BP3 del cromosoma 15q11-q13 y 165 individuos (54.El 5% tenía la deleción típica 15q11-q13 de tipo II, más pequeña, que afectaba a los puntos de rotura BP2 y BP3, y 20 personas tenían una deleción atípica más grande o más pequeña que la deleción típica 15q11-q13 (6,6%). En las personas identificadas con una deleción del cromosoma 15, es importante considerar si una translocación equilibrada podría estar presente en el padre del probando, ya que esto aumenta el riesgo de recurrencia del SPW en la descendencia del padre. Para la disomía materna 15, 185 individuos (36%) tenían disomía uniparental materna 15 (UPD15) con 13 individuos (12,5%) con isodisomía total de todo el cromosoma 15 debido a errores en la meiosis materna II; 60 (57,7%) mostraban isodisomía segmentaria por eventos de cruce en la meiosis materna I y 31 mostraban heterodisomía (29,8%), mientras que en 81 individuos no se realizó el análisis de microarray SNP ni se determinó la clasificación de la disomía materna 15. En cuanto a los defectos de impronta del SPW, se encontraron 22 individuos (4%) con 13 (76,5%) que tenían un estado de epimutación sin deleción, cuatro individuos (23,5%) tenían una microdeleción del centro de impronta mientras que los cinco individuos restantes no tenían un tipo de defecto de impronta establecido. En un estudio relacionado, Hartin et al. (8) llevaron a cabo un análisis más detallado de los defectos de impronta en el SPW utilizando la PCR digital de gota y la secuenciación del exoma completo de próxima generación en una cohorte separada del SPW de 15 pacientes no emparentados y se encontró que dos individuos o el 13% tenían un defecto de microdeleción del centro de impronta. En los 60 individuos con isodisomía segmentaria 15 reportados por Butler et al. (16), el tamaño medio total de la pérdida de heterocigosidad (LOH) fue de 25,1 Mb con un rango de 5-67,4 Mb y un tamaño medio de 16,4 Mb para LOHs individuales. Treinta y dos individuos tenían un segmento de LOH, 25 individuos tenían dos segmentos y tres individuos tenían tres segmentos. Los sitios de LOH más comunes fueron la región proximal 15q11-q13 y la región distal 15q26, incluyendo las bandas 15q12 y 15q26.1 como las más comúnmente registradas.
La presencia de la UPD15 materna y la determinación de la subclase específica (isodisomía segmentaria o total) puede tener un impacto en el diagnóstico y la vigilancia de la atención médica, ya que puede estar presente una segunda condición genética si la madre es portadora de un alelo genético recesivo localizado en la región de LOH que da lugar a dos copias idénticas. En el cromosoma 15 se encuentran cientos de genes potencialmente causantes de enfermedades y éstas deben ser comprobadas o vigiladas de cerca en aquellos con isodisomía segmentaria o total del cromosoma 15. Un diagrama de flujo de pruebas genéticas propuesto para identificar las diferentes clases moleculares tanto para pacientes con SPW como con SA puede verse en el Resumen Gráfico.
Síndrome de Angelman
Se han identificado cuatro clases moleculares reconocidas en el SA que pueden clasificarse por el impacto en la metilación de la región del cromosoma 15. El subtipo más común es una deleción del 15q11 materno.La región 2-q13 es similar a la de origen paterno en el SPW y se encuentra en el ~70% de los individuos con EA (21). Sin embargo, en el SA la deleción típica de clase II es más común. Esta deleción típica de Clase II más pequeña se aproxima más comúnmente a 5 Mb de tamaño de BP2-BP3 y está presente en el 50% de los casos de deleción de AS. Las deleciones de clase I tienen un tamaño de 5-7 Mb y abarcan BP1-BP3 (40% de los casos de deleción). Las deleciones atípicas pueden extenderse desde BP1 o BP2-BP4 o puntos de rotura más distantes. En los individuos con una deleción en la copia materna del cromosoma 15, hay que tener en cuenta si hay signos en el microarray cromosómico que muestren alteraciones que indiquen que podría haber una translocación materna. Esto aumenta el riesgo de recurrencia del SA en la futura descendencia materna. La disomía paterna uniparental 15 representa el 5-7% de los individuos con EA. Los defectos de impronta representan el 3-5% de los individuos con SA y están causados por defectos en el centro de control de la impronta resumidos por Buiting et al. (26). En los individuos con un defecto en el centro de control de la impronta, el marcaje epigenético en la línea germinal no consigue cambiar adecuadamente de un patrón paterno con expresión de UBE3A silenciada para permitir un patrón materno de expresión en el gen UBE3A. Hasta en el 50% de los casos notificados puede identificarse una mutación en el centro de control del imprinting. Se han notificado casos de mosaico de defectos del centro de imprinting en los que un porcentaje de células carece de expresión de la región 15q11.2-q13 y pueden ser más comunes de lo que se pensaba (27). El último defecto genético del SA no afecta a los resultados de las pruebas de metilación del ADN, sino que está causado por una mutación en el gen UBE3A heredado por la madre. Las mutaciones en este gen representan el 11% de los casos de EA (28). Una mutación en el UBE3A podría heredarse por vía materna y, por tanto, está indicado realizar pruebas específicas en la madre del paciente para descartar un riesgo de recurrencia del 50% en su futura descendencia. Si se considera que la mutación es hereditaria, se recomienda considerar la realización de pruebas al abuelo materno de la paciente, ya que esto podría tener implicaciones para los futuros hijos de la tía materna.
Discusión
El manejo médico del SPW y de la EA debe ser dirigido por un equipo multidisciplinar durante la infancia. Tanto los bebés con SPW (más comúnmente) como con SA pueden presentar un retraso en el crecimiento. Un dietista juega un papel importante en el cuidado al principio para tratar el fallo de crecimiento y más tarde en la infancia para evitar la obesidad con la intervención de la dieta con restricción y el uso de programas de ejercicio (que es una preocupación observada más comúnmente para el SPW, pero ahora reconocida en el AS en algunos individuos). Los genetistas clínicos, los especialistas en ortopedia, los médicos de atención primaria, los terapeutas ocupacionales (OT), físicos (PT) y del habla (SLP) especializados, los expertos en salud mental, los especialistas en sueño, los expertos en salud mental y los endocrinólogos son necesarios para abordar los múltiples problemas de salud en el SPW que pueden ocurrir. Un equipo de AS incluye genetistas clínicos, neurólogos, terapeutas especializados para servicios de PT, OT y SLP, especialistas en sueño, gastroenterología, medicina física y rehabilitación, ortopedia y expertos en salud mental. En el caso del SPW, la atención médica adecuada, la gestión y el asesoramiento son los objetivos para controlar el aumento de peso y para controlar y tratar las condiciones comórbidas asociadas, el comportamiento y los problemas psiquiátricos. El crecimiento y otras deficiencias hormonales comunes en este trastorno requieren tratamiento. Un control riguroso de la dieta con seguridad alimentaria y un entorno rutinario controlado con ejercicio regular son estrategias importantes para controlar la hiperfagia, la obesidad y las complicaciones relacionadas que se requieren durante toda la vida. El SA requiere una intervención temprana que incluya el conocimiento de intervenciones terapéuticas especializadas como los dispositivos de comunicación aumentativa y asistencial y un programa de fortalecimiento de ejercicios y actividades de desarrollo intensivo para alcanzar el máximo potencial (por ejemplo, SPIDER), un tratamiento temprano con benzodiacepinas para las convulsiones y una terapia dietética como el uso de una dieta cetogénica. La maximización de todos los aspectos de la atención, incluidos los trastornos del sueño y el estreñimiento, influyen en gran medida en el control de las convulsiones. Un centro especializado y familiarizado con las complejidades y aspectos únicos de estos trastornos puede afectar el resultado.
El diagnóstico temprano es vital para asegurar una intervención temprana tanto para el SPW como para el SA. En el caso del SPW, el diagnóstico precoz debe realizarse durante la infancia para iniciar el tratamiento con la hormona del crecimiento, gestionar los problemas de alimentación, la obesidad, las deficiencias hormonales, los retrasos en el desarrollo y los problemas de comportamiento. El diagnóstico en el SA también asegura las terapias tempranas que impactan en los resultados del desarrollo, así como la profilaxis de las convulsiones, incluyendo la preparación con benzodiacepinas apropiadas. Otras intervenciones que pueden resultar beneficiosas son las dietas especializadas para personas con SA, como la dieta cetogénica o la terapia de bajo índice glucémico (LGIT). El diagnóstico precoz también puede reducir los costes de la atención médica al evitar las hospitalizaciones prolongadas relacionadas con los problemas de alimentación en los individuos con SPW y las convulsiones en los niños con EA.
La identificación de la clase molecular del SPW o del EA con pruebas genéticas avanzadas como los microarrays de SNP de alta resolución permitirá un diagnóstico más preciso, lo que conducirá a mejores indicadores para el pronóstico, y un asesoramiento genético más preciso de los miembros de la familia. Las micromatrices de SNP de alta resolución, el análisis FISH, la amplificación de sondas de ligación específicas de metilación (MS-MLPA) y/o el genotipado del cromosoma 15 son útiles para determinar las deleciones 15q11-q13. Los microarrays de SNP de alta resolución pueden identificar los subtipos de deleción (típica y atípica) tanto en el SPW como en la EA, y las subclases de UPD15 (isodisomía segmentaria e isodisomía total). El subtipo de heterodisomía de UPD y los defectos de CI (microdeleción y epimutación) tanto en el SPW como en el SA pueden requerir un trabajo diagnóstico adicional como se ilustra en el Resumen Gráfico. El subtipo o las clases influyen en el diagnóstico, en el riesgo potencial de recurrencia para los miembros de la familia, en el pronóstico y en el seguimiento de otras condiciones genéticas y características de alto riesgo relacionadas con la clase molecular. Por ejemplo, los rasgos autistas y la psicosis son más comunes en aquellos con SPW y disomía materna 15 y pueden estar relacionados con las subclases específicas de UPD15. Aquellos con las deleciones de clase I más grandes en el SA son más propensos a desarrollar convulsiones difíciles de tratar y microcefalia.
Un diagrama de flujo de pruebas genéticas que incorpora las opciones de pruebas que están disponibles, incluyendo las utilizadas históricamente tanto para el SPW como para el SA, se enumeran en el Resumen Gráfico. Las pruebas para el SPW o el SA a menudo comienzan con la metilación del ADN y, si son anormales, avanzan a otros métodos de pruebas genéticas, incluyendo microarrays SNP de alta resolución o ensayos MS-MLPA basados en la disponibilidad para los médicos y las familias en su entorno clínico. Preferiblemente, se pedirá un array de SNP de alta resolución que esté disponible de forma fácil y comercial en la atención médica occidentalizada. La secuenciación de próxima generación (NGS) del exoma (o del genoma completo) también está disponible para los clínicos, pero la PCR digital de gota (ddPCR) se basa actualmente en la investigación (14). Los arrays de SNP pueden identificar clases moleculares específicas en la mayoría de los pacientes que presentan características del SPW (alrededor del 85% de los casos) o del SA (alrededor del 80%), mientras que el resto de los pacientes necesitarán pruebas adicionales como se describe en el Resumen Gráfico. Pruebas genéticas avanzadas específicas (por ejemplo, ddPCR) pueden ser apropiadamente sensibles para cuantificar el mosaicismo y pueden identificar un diagnóstico en un gran subgrupo de individuos con características clínicas más leves de SPW y AS, pero se necesita más investigación.
Se encontraron diferencias clínicas tempranas al comparar aquellos con SPW o AS que tenían el estado de deleción vs. no deleción (29) incluyendo hipopigmentación en aquellos con SPW y AS que tenían la deleción 15q11-q13 (30). Más tarde, se reportaron mayores puntuaciones de CI verbal (31) o psicosis (32) en aquellos con UPD15 materno comparado con la deleción en individuos con SPW. Además, Butler et al. (19) informaron de puntuaciones de adaptación más bajas y más comportamientos obsesivo-compulsivos en individuos con SPW con la deleción 15q11-q13 Tipo I en comparación con la UPD15. Zarcone et al. (33) informaron que los individuos con SPW y la deleción 15q11-q13 Tipo I tenían más compulsiones con el aseo personal y comportamientos compulsivos difíciles de interrumpir y que interferían con las actividades sociales más que aquellos con deleciones Tipo II o UPD15. En un estudio de correlación fenotipo-genotipo en el síndrome de Angelman, Moncla et al. (34) informaron de un aumento de la actividad convulsiva en aquellos con la deleción de clase I más grande en comparación con los que no tenían deleción. La microcefalia, la ataxia, la hipotonía y las dificultades de alimentación también son más probables en el subtipo de deleción (3). Pueden presentar un deterioro del lenguaje más grave, en particular del lenguaje receptivo y rasgos autistas (21, 35). Los individuos con EA con UPD paterna pueden tener un mejor lenguaje receptivo, mejores habilidades motoras y una menor prevalencia de convulsiones. Los individuos con mosaico también pueden tener un fenotipo más leve que incluye mejores habilidades lingüísticas, funcionamiento adaptativo y menos convulsiones (36).
La secuenciación del exoma de próxima generación o del genoma completo también puede tener un lugar en las evaluaciones genéticas del SPW o del SA, particularmente en aquellos individuos que presentan hallazgos inusuales o un diagnóstico tardío (por ejemplo, UPD15 segmentaria o isodisomía total) y en los casos en los que el ADN paterno no está disponible (8). Para abordar el uso y el tipo de pruebas genéticas para el SPW y el SA, se desarrolló un nuevo diagrama de flujo de pruebas genéticas para el clínico como se describe e ilustra en el Resumen Gráfico. Este diagrama de flujo puede ayudar a pedir pruebas genéticas basadas en la presentación clínica para determinar el diagnóstico, el manejo y el tratamiento apropiados y para proporcionar la información más precisa de asesoramiento genético para otros miembros de la familia. Sugerimos el uso de este algoritmo para completar definitivamente el trabajo de diagnóstico tanto para el SPW como para el SA. Argumentamos que el diagnóstico es incompleto sin el conocimiento del subtipo genético específico del paciente para guiar el asesoramiento, la orientación anticipada, el manejo y las probables opciones terapéuticas. La determinación de la clase molecular es importante para la atención médica y el tratamiento y útil para el clínico comprometido en el asesoramiento genético de los miembros de la familia para el SPW o AS.
Contribuciones de los autores
MB y JD contribuyeron a la redacción del manuscrito, la revisión de la literatura, contribuyeron con su experiencia y editaron el manuscrito.
Financiación
Reconocemos la subvención del Instituto Nacional de Salud Infantil y Desarrollo Humano número HD02528.
Conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un potencial conflicto de intereses.
Agradecimientos
Reconocemos a Grace Graham por la preparación experta del manuscrito.
1. Bittel DC, Butler MG. Síndrome de Prader-Willi: genética clínica, citogenética y biología molecular. Expert Rev Mol Med. (2005) 25:1-20. doi: 10.1017/S1462399405009531
CrossRef Full Text | Google Scholar
2. Butler MG. Butler MG, Lee PDK, Whitman BY. Management of Prader-Willi Syndrome. New York, NY: Springer. (2006). doi: 10.1007/978-0-387-33536-0
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Williams CA, Driscoll DJ, Dagli AI. Aspectos clínicos y genéticos del síndrome de Angelman. Genet Med. (2010) 12:385-95. doi: 10.1097/GIM.0b013e3181def138
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Cassidy SB, Schwartz S, Miller JL, Driscol DJ. Prader-Willi syndrome. Genet Med. (2012) 14:10-26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Angulo MA, Butler MG, Cataletto ME. Síndrome de Prader-Willi: una revisión de los hallazgos clínicos, genéticos y endocrinos. J Endocrinol Invest. (2015) 38:1249-63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Butler MG. Causas monogénicas y sindrómicas de la obesidad: ejemplos ilustrativos. Prog Mol Biol Transl Sci. (2016) 140:1-45. doi: 10.1016/bs.pmbts.2015.12.003
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Imprinting genético sugerido por la heterodisomía materna en el síndrome de Prader-Willi sin deleción. Nature. (1989) 342:281-5. doi: 10.1038/342281a0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hartin SN, Hossain WA, Francis D, Godler DE, Barkataki S, Butler MG. Análisis del centro de impresión del síndrome de Prader-Willi utilizando PCR digital de gota y secuenciación del exoma completo de próxima generación. Mol Genet Genomic Med. (2019) 7:e00575. doi: 10.1002/mgg3.575
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hartin S, Hossain WA, Weisensel N, Butler MG. Tres hermanos con síndrome de Prader-Willi causado por microdeleciones del centro de impresión y revisión. Am J Med Genet A. (2018) 176:886-95. doi: 10.1002/ajmg.a.38627
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Hassan M, Butler MG. Prader-Willi syndrome and atypical submicroscopic 15q11-q13 deletions with or without imprinting defects. Eur J Med Genet. (2016) 59:584-9. doi: 10.1016/j.ejmg.2016.09.017
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Fountain MD, Schaaf CP. Síndrome de Prader-Willi y síndrome de Schaaf-Yang: enfermedades del neurodesarrollo que se cruzan en el gen MAGEL2. Diseases. (2016) 13:4. doi: 10.3390/diseases4010002
CrossRef Full Text | Google Scholar
12. Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Fenotipo de Prader-Willi causado por la deficiencia paterna para el cluster de ARN nucleolar pequeño HBII-85 C/D box. Nat Genet. (2008) 40:719-21. doi: 10.1038/ng.158
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Rhyman DC, et al. Prader-Willi-Like phenotype caused by an atypical 15q11.2 microdeletion. Genes (Basilea). (2020) 25:11. doi: 10.3390/genes11020128
CrossRef Full Text | Google Scholar
14. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawfrd JD. Deleciones del cromosoma 15 como causa del síndrome de Prader-Willi. N Engl J Med. (1981) 304:325-9. doi: 10.1056/NEJM198102053040604
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet. (1983) 4:1285-6. doi: 10.1016/S0140-6736(83)92745-9
CrossRef Full Text | Google Scholar
16. Butler MG, Hartin SN, Hossain WA, Manzardo AM, Kimonis V, Dykens E. Clasificación genética molecular en el síndrome de Prader-Willi: un estudio de cohorte multisitio. J Med Genet. (2019) 56:149-53. doi: 10.1136/jmedgenet-2018-105301
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Butler MG, Thompson T. Síndrome de Prader-Willi: hallazgos clínicos y genéticos. Endocrinologist. (2000) 10:3s-16s. doi: 10.1097/00019616-200010041-00002
CrossRef Full Text | Google Scholar
18. Prader-Willi. Butler MG. Síndrome de Prader-Willi: conocimiento actual de la causa y el diagnóstico. Am J Med Genet. (1990) 35:319-32. doi: 10.1002/ajmg.1320350306
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Diferencias conductuales entre sujetos con síndrome de Prader-Willi y deleción tipo I o tipo II y disomía materna. Pediatrics. (2004) 113:565-73. doi: 10.1542/peds.113.3.565
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Fases nutricionales en el síndrome de Prader-Willi. Am J Med Genet A. (2011) 155:1040-9. doi: 10.1002/ajmg.a.33951
CrossRef Full Text | Google Scholar
21. Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, et al. Distintos fenotipos distinguen las clases moleculares del síndrome de Angelman. J Med Genet. (2001) 38:834-45. doi: 10.1136/jmg.38.12.834
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Trickett J, Oliver C, Heald M, Denyer H, Surtess A, Clarkson E, et al. Evaluación multimétodo del sueño en niños con síndrome de Angelman: un estudio de casos controlados. Front Psychiatry. (2019) 10:874. doi: 10.3389/fpsyt.2019.00874
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Buiting K, Clayton-Smith J, Driscoll DJ, Gillessen-Kaesback G, Kanber D, Schwinger E, et al. Tarjeta genética de utilidad clínica para: Angleman syndrome. Eur J Hum Genet. (2015) 23:2. doi: 10.1038/ejhg.2014.93
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. (2008) 4:577-84. doi: 10.2147/NDT.S2749
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Bindels-de Heus KGCB, Mous SE, Hooven-Radstaake MT, van Iperen-Kolk B, Navis C, Rietman AB, et al. Una visión general de los problemas de salud y desarrollo en una gran cohorte clínica de niños con síndrome de Angelman. Am J Med Genet A. (2020) 182:53-63. doi: 10.1002/ajmg.a.61382
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Buiting K, Williams C, Horsthemke B. Angelman syndrome – insights into a rare neurogenetic disorder. Nat Rev Neurol. (2016) 12:584-93. doi: 10.1038/nrneurol.2016.133
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, et al. Síndrome de Angelman atípico debido a un defecto de impresión en mosaico: informes de casos y revisión de la literatura. Am J Med Genet A. (2017) 173:753-7. doi: 10.1002/ajmg.a.38072
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics. (2015) 12:641-50. doi: 10.1007/s13311-015-0361-y
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Butler MG, Meaney FJ, Palmer CG. Estudio clínico y citogénico de 39 individuos con síndrome de Prader-Labhart-Willi. Am J Med Genet. (1986) 23:793-809. doi: 10.1002/ajmg.1320230307
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Butler MG. Butler MG. Hipopigmentación: una característica común del síndrome de Prader-Labhart-Willi. Am J Hum Genet. (1989) 45:140-146.
PubMed Abstract | Google Scholar
31. Roof E, Stone W, MacLean L, Feurer ID, Thompson T, Butler MG. Intellectucal characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res. (2000) 44(Pt 1):25-30. doi: 10.1046/j.1365-2788.2000.00250.x
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. (2002) 359:135-6. doi: 10.1016/S0140-6736(02)07340-3
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S, Caruso-Anderson M, et al. La relación entre el comportamiento compulsivo y el rendimiento académico a través de los tres subtipos genéticos del síndrome de Prader-Willi. J Intellect Disabil Res. (2007) 51:478-87. doi: 10.1111/j.1365-2788.2006.00916.x
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Moncla A, Malzac P, Voelckel MA, Auquier P, Girardot L, Mattei MG, et al. Correlación fenotipo-genotipo en 20 pacientes con síndrome de Angelman con deleción y 20 sin deleción. Eur J Hum Genet. (1999) 7:131-9. doi: 10.1038/sj.ejhg.5200258
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, et al. Identificación de nuevas deleciones de 15q11q13 en el síndrome de Angelman por array-CGH: caracterización molecular y correlaciones genotipo-fenotipo. Eur J Hum Genet. (2007) 15:943-9. doi: 10.1038/sj.ejhg.5201859
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preservación del lenguaje expresivo como determinante fenotípico del síndrome de Angelman mosaico. Mol Genet Genomic Med. (2019) 1:e837. doi: 10.1002/mgg3.837
CrossRef Full Text | Google Scholar